Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
Chapter 26, Exploring with HSP – (Generating and testing research hypotheses)
But HSP can also be used by the adventurous
explorer to map out uncharted territory. All that’s needed is a bunch of HSP
values, a vague hypothesis and some extra support software such as Excel.
Mission
impossible – the insoluble polymer
Charles once had to find the best
high-temperature solvent for a polymer that was insoluble in just about
everything at room temperature. By chance it was known that one horribly toxic
solvent could just about dissolve the polymer at ~150ºC –
which was not a practical temperature. Knowing its HSP it was then possible to
look for other solvents with HSP in that sort of region – just by sorting
with RED number. This give a short list of possible candidates.
These were tested. Not surprisingly, given
the single datapoint that started this process, about half the solvents were
worse than the toxic one, but some were better. From this small dataset a
better HSP estimate could be made and a few more solvents could be tested. This
then gave a practical solvent that worked at ~120ºC.
That’s a
simple example of exploration. The original hypothesis wasn’t brilliant. But it
was a start. Without that hypothesis the solvent screening process would have
involved many more experiments and much more time and expense. Now let’s look
at some more complex explorations.
Screening
with Distance maps
Suppose, (to take a specific example from
the HSP user who inspired this chapter) you are interested in plant chemicals.
You have an idea that solubility plays a key part (“a necessary but not sufficient
condition”) in allowing a chemical to reach its target as, say, an
anti-malarial. There are innumerable plant chemicals out there and if you don’t
have access to a pharma-grade high-throughput screening system, how do you
narrow down your choices?
The key is to have a method not of hitting
winners (that’s asking too much) but excluding no-hopers. The problem with
screening is that there are far too many possible candidate molecules, so
anything which has a reasonable chance of excluding molecules that won’t work
will be of great help.
Your starting point is a (small) list of
chemicals that are known to work. If they have very different HSP then you need
not bother to continue. But if (as is often the case) they are clustered near
one region of HSP space then you can create a “target” value from this cluster
(e.g. using a Sphere, taking an average, choosing your favourite), ready for
calculating the distances from this target of the pre-screen molecules. An
example of this approach has already been described in the DNA chapter with the
cytotoxic chemicals.
So assemble your list of plant chemicals as
a list of Names and SMILES and save it as a simple tab-separared .txt file
– Excel does this for you no problem. Then find
the File Convert option in Y-MB. If you have a lot of molecules and many of
them are large you may want to go and have a cup of coffee while the computer
does all the work. At the end of the process you have a .hsd file with
estimated HSP values for most of the chemicals you presented. Most? It’s likely
that any list of SMILES will have a few problem chemicals which Y-MB can’t
handle. But if you drag the .hsd file into Excel it’s smart enough to recognise
that it’s a tab-separated format dataset and you’ll get a nice table. Search
for the word “error” using Excel and you’ll find the failed molecules. Simply
delete their rows – you’ve got more than enough chemicals to screen in
any case – or find the correct SMILES, convert them manually in Y-MB and
Paste the values into Excel.
Now create a fresh line at the top which
contains the target value which you think (or hypothesise) is a fair
representation of the class. Then it’s easy to calculate the HSP distance of
each molecule from your target D=sqrt(4*(δDt-δDi)2
+ (δPt-δPi)2 + (δHt-δHi)2)
where t=target and i=the i’th chemical. Excel can then sort by the distance
column and you can decide a cut-off value for screening purposes –
rejecting anything with a distance greater than that value.
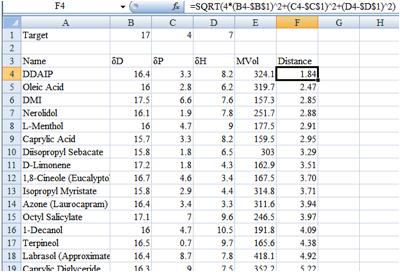
Figure 1‑1 A distance map in Excel, sorted to show the chemicals closest to
the target
In this dummy example (of course there are
more chemicals not included in the screen shot) I might decide that anything
less than a distance of 4 is acceptable so would only test DDAIP to Octyl
Salicylate.
If you want to be more sophisticated you
can reason that anything with a MVol > 400 (or whatever value you choose)
will be too slow to penetrate the target or too hydrophobic (see the chapter
describing Ruelle’s solubility calculations) so you can do a sub-sort and
remove those molecules that are too large. For this example I sub-sorted the
<4 area by MVol, then resorted the area with MVol<300 to give my final
shortlist:
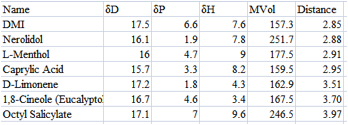
Figure 1‑2 A smaller list by excluding high MVol chemicals
If you’ve already got your list of
pre-screening molecules in SMILES format then this whole exploration will have
taken less than a few hours. What will you have achieved? At the very least,
the molecules that pass your distance screen will be (because of their HSP
match) compatible with the
formulation vehicle of your target active. If you’re an optimist then there’s
also a good chance that these molecules will partition into the correct part of
the cell or organism (because of the HSP match) and have a chance of working.
When we’ve tried such explorations we find
them to be much more insightful than methods typically used in pharma:
Lipinski’s Rule of 5 or LogP. No simple method will ever be perfect for identifying
candidates but we believe that the HSP distance method has a lot going for it.
Finally, remember that there’s more to
efficacy than solubility! It’s up to you to sub-screen the molecules for the
right sort of chemical functionality. An example can be drawn from the
chemotherapy drugs mentioned above. In the specific case of carboplatin, it is
the organic segment of the drug that has the proper HSP, and not the platinum,
but the drug can orient such that the platinum is hidden within a kind of micelle,
thus allowing the desired effect.
SOMs
Very often the field of exploration isn’t a
simple yes/no – good drug/ bad drug. There may be a set of
characteristics which you need to map to find if there is a link with the 3D
space of HSP. Those who are skilled in Principle Component Analysis can readily
take a large HSP dataset and try to map the δD, δP, δH and
(usually) MVol values against the factors of interest. But we find that SOMs,
Self Organising Maps, provide a better view of complex terrain. They attempt to
put “like-with-like” within a 2D space and the hope is that there will be
clear-cut regions on the map that distinguish one type of behaviour from
another. If you are not familiar with SOMs then the Wikipedia article is a good
introduction.
A simple example is the Sphere technique
itself. On a SOM a good Sphere fit is equivalent to a (roughly) circular area
of “good” molecules all separated from the rest of space which contains the
“bad” molecules. In Hiroshi’s work on the new fitting regimes (Double-Sphere,
Data…) he found that SOMs were very helpful in revealing problems with the
datasets.
Here is an attempt to make the data fit a
double sphere SOM. The lines are “guides for the eye” and not part of the SOM
software output:
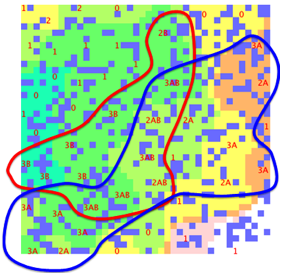
Figure 1‑3 A double-sphere SOM
Within this eBook we’ve already mentioned
SOMs in terms of fragrance mapping. Such maps aren’t definitive about cause and
effect, but they are suggestive of
hypotheses which can be further explored.
Fitting
Another example from this eBook illustrates
another way to explore. We were interested in the partition coefficient between
soil and water, Koc. It seemed reasonable to guess that the HSP
distance between the chemical and some hypothetical soil would correlate with
the partition coefficient. But what is the HSP of soil? The answer was to take
a set of known Koc values, make a guess at the HSP of soil,
calculate the distances from each chemical and that soil and, by adding some
further coefficients, predict Koc from the distance. The square of
the errors between predicted and actual values could then be summed –
showing that the original guess, not surprisingly, was hopelessly wrong.
That’s when Excel comes in. Ask its Solver
to minimise the error sum by varying the coefficients and the HSP of soil and
see what happens. If the fit is still bad then the hypothesis is useless. If
the fit is good then you have an effective working HSP for soil and can play
around with some extra parameters (MVol or MWt usually have a part to play) to
find the best fit with the least adjustable values.
If you have more sophisticated fitting
algorithms then you can automatically find complex relationships which will
give even better fits.
Once you’ve got some good fits it’s time to
pause. Do the fits make sense in terms of basic chemistry and thermodynamics?
Have you just found some meaningless relationship by providing too many fitting
parameters, or does the relationship suggest some interesting science? Do the
fits from the test data make good predictions for data not included in the
fitting set?
The answers to those questions will vary.
Sometimes the fits really are artificial and tell you nothing. Sometimes they
fit so beautifully to standard HSP theory that you can be pretty sure that they
are sound. But remember that the fits aren’t an end in themselves – they
are a means to deeper understanding and, perhaps, useful predictions.
To
boldly go
With the tools provided by HSPiP and with
the extra tools in Excel (and other programs) it’s not hard to explore. Often,
as with real explorers, the results are failures. But when the risks are small
and the rewards are significant, maybe it’s worth having a go. We’ve used
HSPiP’s tools for our own explorations and so made them as easy as possible to
use. We hope you will want to use them for your own journeys into the unknown.
E-Book contents | HSP User's Forum
