Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
Chapter 30, DIY HSP (Methods to Calculate/Estimate Your Own HSP)
So it would be very nice if there were a
universally validated method for calculating HSP to a reasonable degree of
accuracy. Unfortunately some of the methods require knowledge of other values
such as enthalpy of vaporization or dipole moment and you may not know either
or both of those.
The recent advances in statistical thermodynamics
by Panayiotou and others offer some encouragement that HSP calculations will
become more accurate and more routine in the years ahead. Similarly, work on
molecular dynamics has proven fruitful in calculating δTot and recent work by,
e.g. Goddard’s group in CalTech has shown tantalising evidence of being able to
calculate δD, δP and δH. However, all these techniques are still only do-able
in the hands of expert teams. So in the meantime we have to use a variety of
approaches and, most importantly, our own judgement.
The most basic calculation is of δTot. This
is simple:
δTot = (Energy of Vaporization/Molar
Volume) ½
But where do you find your energy (enthalpy
– RT) and your molar volume? There are extensive tables of enthalpy
values available at a price. Any modern molecular mechanics program can do a
reasonable job of calculating molar volume and there are also free on-line
tools.
So you might be lucky and be able to
calculate δTot.
δP has been shown to be reasonably
approximated by the simple Beerbower formula which requires just one unknown,
the dipole moment:
δP=37.4 * Dipole Moment/MVol ½
The more complex Böttcher equation (see
equation 10.25 in the Handbook) requires
you to know the dielectric constant and refractive index in addition to the dipole
moment. It may arguably give better values if you have accurate values for all
the inputs, but it is unlikely that you have those inputs so you are no better
off.
The correlation has been re-done using the
updated HSP list which, in turn was updated on the basis of the most recent
databases of dipole moments. There is a necessary circularity to this process
but the aim is self-consistency with all
available experimental data so the process is highly constrained. The new fit,
based on 633 values is shown in the graph:
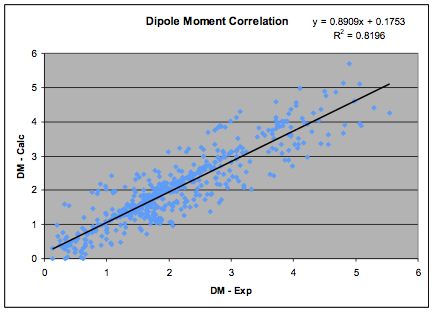
Figure 1‑1 The Dipole Moment correlation
and the revised formula, which is used in
the HSPiP software is:
δP=36.1 * Dipole Moment/MVol ½
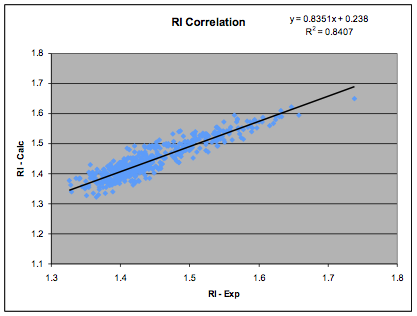
The paper by D.M. Koenhen and C. A. Smolders, The Determination of Solubility Parameters
of Solvents and Polymers by Means of Correlations with Other Physical
Quantities, Journal Of Applied Polymer Science 1975, 19, 1163-1179 does
what the title suggests and finds not only an acceptable equivalent to
Beerbower (they had an alternative power dependency for MVol but our revised
data confirmed that 0.5 is optimal) but also a simple linear relationship
between δD and refractive index. The coefficients shown here are our own fit to
a more extensive and revised data set of 540 data points:
Figure 1‑2 The RI correlation
δD= (RI - 0.784) / 0.0395
Koenhen and Smolders also showed a strong
correlation between δD2+δP2, MVol0.33 and
surface tension. Using 498 data points with relevant surface tension data we
found a correlation:
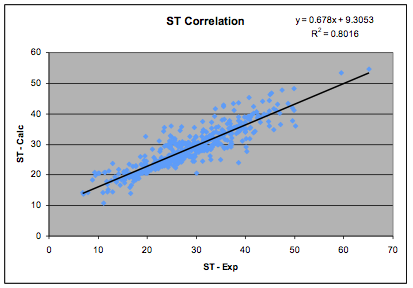
SurfTension=0.0146*(2.28*δD2 + δP2 + δH2)*MVol0.2
Figure 1‑3 The Surface Tension correlation
When it comes to δH there is no obvious
short-cut for calculating it from first principles using a few constants. We
therefore have to rely on group contribution methods. And because we can use
such methods for those, we might as well try to use them for δTot, δD and δP as
well.
Group
contributions
There is a long and distinguished history
of breaking molecules down into a number of smaller sub-groups then calculating
a property by adding together numbers for each group, weighted by the numbers
of such groups in the molecule. There is an obvious trade-off in group
contributions. It’s possible to define –CH2- as just one group or as 2
groups (-CH2- in acyclic and in cyclic molecules) or many groups (-CH2- in
acyclic, in 3-member rings, in 4-member rings in 5-member rings etc. etc.
etc.). The more subgroups used the more accurate, in principle, the group
contribution but the less likely that there is sufficient statistical data to
calculate the fits with any degree of reliability.
Over the years there has been a convergence
on the so-called UNIFAC partition of groups – providing an adequate
balance between over-simplification and over-complication.
So to calculate the group contributions for
D, P and H one “simply” divides a set of molecules with known HSP into their individual
groups then does a linear regression fit to the data at hand. In practice this
is a lot of work and only a few such fits exist for HSP.
Because δD comes from Van der Waals forces
it is intuitively obvious that group contribution methods should produce
reasonable approximations. It doesn’t matter all that much where a C-Cl bond is as the more important fact is
that there is both a C and a Cl.
δP is obviously problematic. A molecule
with two polar groups near one end is likely to be more polar than one where
those two groups are at opposite ends and tend to cancel out. It is hard for a
group method to capture the geometrical issues.
Similarly, it is obvious that molecules
with two –OH groups in them might differ strikingly in the amount of
hydrogen bonding interactions between
molecules depending on how much hydrogen bonding there is within each molecule. So δH can never be accurately determined from
group methods.
So no matter how hard you try, you can’t
realistically expect always to get accurate δH and δP values from group
methods.
How
does Hansen do it?
The overall goal is to divide the cohesion
energy (Energy of Vaporization) into the three parameters discussed above. One
finds or estimates the latent heat of vaporization at 25°C and subtracts RT (395
cal/mol or 1650 J/mol).
The preferred method to find δD is to use
one of the figures in Chapter 1 of the Handbook.
These give the dispersion energy of cohesion as a function of the molar volume.
There are curves for different reduced temperatures. Use of reduced
temperatures is characteristic of a corresponding states theory, which means
that the HSP are based on corresponding states. The reduced temperature is
298.15/Tc. Tc is the critical temperature that can be
found for many (smaller) molecules, but not for the larger ones. This then
requires estimation. The Tc has been estimated by the Lydersen
method as described in the Handbook
using group contributions from the table for this purpose. Tc is
found by dividing the temperature at the normal boiling point by the constant
found from the Lydersen group contribution. One can then easily find δD with
this energy and the molar volume. When this preferred procedure is not possible
one can compare with similar compounds. Remember that δD increases with
molecular size, especially for aliphatic molecules. This combined with the
probable incompatibility of group contributions with a corresponding states
theory makes the accurate estimation of δD especially difficult, especially for
polymers.
δP is usually found with the Beerbower
equation given in the above, or else by group contributions as reported in the Handbook. If a dipole moment can be
found for a closely related compound, its δP can be found with its molar
volume, and this can then be used to find a new group contribution value for
use with the table in the Handbook.
This procedure is best when the whole procedure of finding HSP values is
possible for the related compound. The ultimate result is two new sets of HSP.
δH in the earliest work was found
exclusively by difference. The polar and dispersion cohesion energies were
subtracted from the total cohesion energy to find that left over. This was then
used to find δH. When things did not add up properly comprises were made based
on the multitude of experimental data that were generated in the process of
establishing the first values. Up to the point where the Panayiotou procedure
came forth, the usual method of estimating δH was with group contributions as
given in the Handbook.
For the sake of historical record, note
that the original values reported by Hansen in 1967 were expanded to a total of
about 240 by Beerbower using the Böttcher equation, his own equation, and
the group contributions in the tables in the Handbook. This set was then extended over the years by one method
or another by Hansen to arrive at the values found in the Handbook. The revision process will presumably continue, but the
original values from 1967 seem to be holding up well. The Hoy parameters are
not compatible with the Hansen parameters, particularly with respect to finding
a dispersion parameter that is too low. The Van Kevelen procedure also gave
somewhat inconsistent values and did not have a wide selection of groups to
use. Experimental data were found where possible and practical, and adjustments
made accordingly, but one must do this with care, since what looks good in one
correlation may totally ruin another.
Much
better than nothing
So now you know the bad news about DIY HSP.
There is currently no good way to be sure you have calculated accurate values,
for reasons which are fundamental. So, do we abandon hope?
Happily the answer is that by using all the
available methods and combining them with your own scientific understanding,
it’s possible to get HSP that are fit for purpose. If the molecule is well
outside the sphere it doesn’t really matter how far outside it is. So it often
doesn’t matter if δH is 12 or 14. It suffices for you to know that it’s not 2
or 4.
And if you find that it’s critical to know
if δH is 12 or 14 so that you can really refine the radius of the Sphere, you
can resort to good old-fashioned experiment to get the HSP for the one molecule
that happens to be of critical importance.
The 6
ways
In the program we offer you 6 ways to
calculate HSP.
1 This lets you input enthalpy, molar
volume, refractive index and dipole moment. You therefore get δTot and δP. If
you also enter an estimate for δD the program calculates δH. You can also
correct for temperature of calculation of enthalpy and see an estimation of the
surface tension from your calculated parameters.
2 The most extensive and accurate published
group-contribution method for all 4 values (δTot, δD, δP and δH) comes from
Panayiotou’s group in Aristotle University in Thessaloniki. The Stefanis-Panayiotou
method (E. Stefanis, C.Panayiotou, Prediction
of Hansen Solubility Parameters with a New Group-Contribution Method,
International Journal of Thermophysics, 2008, 29 (2), 568-585) has established
itself as an important method. The extra feature of S-P is that it attempts to
distinguish different forms of similar groups by identifying 2nd-order
groups which have their own parameters. If you want a rough estimate, then keep
things simple and ignore the 2nd-order groups. For more accuracy you
must include the 2nd-order groups. It can be difficult to know how
to partition your molecules into these UNIFAC groups. Helpfully, S-P provided
an example of each type of 1st- and 2nd-order group to
help you break down your molecule in the correct manner.
A typical example is 1-Butanol which has 1
CH3- group, 3 –CH2- groups and one –OH group. If you enter these (1st-order)
values and press calculate you get values for δTot and then δD, δP, δH of 21.9
and [15.9, 5.9, 13.2] (c.f. [16, 5.7, 15.8]) respectively.
There is a further refinement. If you are
confident that the molecule (for whatever reason) will tend to be of low δP
and/or δH, you can click the “Low” option and use group parameters tuned for
these respective properties. To help you with your intuition, if you attempt,
for example, to use the Low H option for 1-Butanol you get a warning because
there is not (should not be!) a Low H fitting parameter for this molecule.
For users who aren’t too comfortable in
creating the UNIFAC groups, the Y-MB method below provides an automatic way of
creating these groups (first-order only) from Smiles or 3D molecule input. No
automatic group method can be 100% accurate so you need to do your own sanity
check, but in our tests it has proven to be most helpful. It’s also insightful
to compare the HSP predictions of the two methods – they both have their
strengths and limitations.
3 Van Krevelen is the first to admit that
his group method cannot give accurate results, for the reasons discussed above.
His particular contribution to the problem is to introduce a “symmetry” option.
If there is one plane of symmetry then the polar value is halved, with two
planes it is quartered and with 3 planes both the polar and hydrogen bonding
values are set to zero. The one-plane choice, for example, would help
distinguish our two cases of C-Cl bonds discussed above.
4 Hoy uses a more subtle form of
calculation from his chosen groups and includes options similar to Panayiotou’s
secondary groups by taking into account various x-membered rings and some forms
of isomerism. Importantly, Hoy also attempts to make corrections for polymers.
It’s intuitively obvious that, for example, the polar effect of an isolated
sub-unit would be rather different from the overall polar effect from the
polymer chain made up from those sub-units.
Hoy also helps with input to the numerical
and Van Krevelen calculations by producing an approximate value for the molar
volume. This can’t be as accurate as a proper measurement from density and molecular
weight or from a molecular mechanics program, but it’s a useful aid if you
can’t derive it from those sources.
5 One of the issues with group methods is
that they often can’t satisfactorily predict complex inter-group interactions.
Hiroshi Yamamoto therefore adapted his Neural Network (NN) methodology for
fitting the full HSP data set in
such a way that inter-group interactions automatically get fitted by the
relative strengths of the neural interconnections. But of course this needed
him to have a set of groups. He therefore devised an automatic Molecule
Breaking (MB) program that created sub-groups from any molecules. He used a
general MB technique that allowed him to experiment with which combination of
MB and NN gave the best predictive power for HSP. That’s what you get with
HSPiP. And because the MB technique was general, he was able to take standard
molecular inputs (such as Smiles, MolFile (.mol and .mol2), PDB and XYZ) and
"break" them so the user can get completely automatic calculation of
individual molecules (plus their formula and MWt) or, given a table of Smiles
chemicals, bulk conversion to a standard .ssd file with a large set of
chemicals. [If you happen to have a set of chemicals in another format, such as
Z-matrix, which HSPiP cannot handle, then we recommend OpenBabel, the Open
Source program that provides file format interconversion for just about
anything that’s out there. We used OpenBabel a lot when we were developing the
implementation of Hiroshi’s technology]. Charles and Steve have called the
method Y-MB for Yamamoto Molecular Breaking [Hiroshi was too modest to want
such a name] and we believe that Y-MB represents a fundamental change in the
way HSP can be used in the future. Hiroshi’s extensive knowledge of Molecular
Orbital (MO) calculations and their interpretation means that in the future
Y-MB might be augmented via MO.
In addition to the HSP values, Y-MB
provides estimates of many other important parameters such as MPt, BPt, vapour
pressures, critical constants and Environmental values.
Like all group contribution methods, Y-MB
isn’t magic. It can’t accurately predict values for groups or arrangements of
groups that are not in its original database. The more HSP that can be measured
independently, the more Hiroshi can refine the Y-MB technique to give better
predictions. As mentioned above, the Y-MB breaking routine can optionally find
the Stefanis-Panayiotou UNIFAC groups.
For the 3rd Edition, Hiroshi
carried out a huge analysis of results on a database of many thousands of
molecules including many pharma, cosmetic and fragrance chemicals. From this he
was able to refine his list of group fragments and also test novel NN and
Multiple Regression (MR) fits. As a result we now have internal NN and MR
variants for calculating the different parameters of Y-MB. Each has its own
strengths for different properties. For the user the only difference from
previous editions is that the estimates are often improved – particularly
for very large molecules where we acknowledged that the original Y-MB had
problems.
Because we believe that the relatively new
InChI (International Chemical Identifier) standard for describing molecules is going to be of
great future importance, we output the “standard” InChI and InChIKey.
These are created with the “No Stereochemistry” option so they are the simplest
possible outputs. Importantly, if you use the first 14 digits of the InChIKey
as the search string on places such as ChemSpider (probably the best
one-stop-shop for information on a chemical) then you are guaranteed to get the
correct matches. InChIKeys are unique identifiers created from the InChI so
unlike CAS# they are directly traceable to specific molecules and there is only
one InChIKey (well, the first 14 digits) to a molecule. The reason we emphasise
the first 14 digits is that they will find all variants of a given molecule,
independent of stereochemistry, isotope substitution etc. Once you start using
InChIKeys for searches you’ll wonder how you ever survived without them.
For a useful quick guide to InChI, visit http://en.wikipedia.org/wiki/International_Chemical_Identifier
6 Polymers are a problem. We have no
reliable general method for predicting polymer HSP. This is not surprising. For
example, there is no such thing as “polyethylene”; instead there are many
different “polyethylenes” and it would be surprising if their HSP were all
identical. But that doesn’t mean that we should give up. An intelligent
estimate can often provide a lot of insight. Hiroshi had proposed an extension
of his Y-MB technique to include polymers. And by good fortune we found Dr W.
Michael Brown’s website at Sandia National Laboratories:
http://www.cs.sandia.gov/~wmbrown/datasets/poly.htm
With great generosity, Dr Brown gave us
permission to use his dataset. Hiroshi then implemented a revised version increasing
the number of polymers from <300 to >600. To make it more consistent with
the rest of the program we’ve used –X bonds as symbols of the polymer
chain rather than the pseudo-cyclic “0” used by Dr Brown.
To calculate the polymer HSP, simply double
click (or Alt-Click) on one of the polymers. This puts the Smiles up into the
top box. Then click the Calculate button as normal. You can, of course, enter
your own polymer Smiles manually if you wish.
As the whole area of polymer HSP prediction
is so new, the Y-MB values for a single monomer repeat can often be somewhat
unreliable. You can, therefore, set a number of repeating units, say, 4, and
the full polymer Smiles for this 4-mer is created and the Y-MB values
calculated. You can use your own judgement as to which value to use – the
1-mer, 2-mer, 3-mer … There are some complications to this automated process.
If, for example, you had a 2-ring monomer and asked for a 5-mer, you will get a
message to say that this is impossible – the problem is that the first
rings would be labelled 1,2, the second 3,4 and the 5th repeat unit
would be 9,10 – and polymer Smiles can only use rings from 1-9.
Although this is hugely helpful, we think
there’s even more that can be done with this. With a bit of intelligent
copy/paste you can construct polymer blends. For example, if you take
polyethylene, C0C0, and polycyclohexylethylene, C0C0C2CCCCC2, you can combine
them to create the ABAB copolymer C0CCC0C2CCCCC2, or the AABBAABB copolymer
C0CCC CC(C2CCCCC2)CC0C2CCCCC2 etc. It’s a bit tricky (note the extra
parentheses around the middle cyclohexyl group) but it’s pretty powerful. To
help you we’ve added a CP
(Co-Polymer) button that you can click when you’ve selected two polymers from
the database. The program automatically creates an AB, AABB or AAABBB polymer
according to your choice. Note that it is possible to make “impossible”
polymers this way – the program makes no effort to see if two monomers
could actually be made into a co-polymer.
Again we need to stress that this is all so
new that the predicted values should be treated with caution. Above all we need
many more experimental data points for polymers and it seems that IGC offers a
lot of hope for the routine gathering of a lot of relevant data. Armed with
more data, the polymer Smiles predictions can be refined.
We had pointed out to users of the Polymer
Smiles method in earlier editions that the limitations were significant. With
the improved Y-MB version we are much happier that Polymer Smiles are more
stable and insightful. They should still be used with caution, but their
capabilities are clearly much improved for the 3rd Edition.
If you want to see the structure of any of
the polymers, Ctrl-Shift-click on the polymer in the database and a 3D
representation appears in the Y-MB tab. We created the 3D structures automatically
from the polymer Smiles using the public domain OpenBabel utility.
Revisions
to the HSP table
We’ve used all the above considerations to
update the HSP data used in HSPiP. Many of the changes have been minor, some
will be more significant. Any changes will be unwelcome to those who have been
using the Hansen table for years. So it’s worth explaining why we made the
changes.
There is a fundamental principle that all
worthwhile databases contain errors. The published Hansen table contained a few
typos, and a few errors. But many of the changes have come about because the
basic data in other databases such as DIPPR
801 and Yaws' Handbook of
Thermodynamic and Physical Properties of Chemical Compounds have changed.
Thanks to Hiroshi Yamamoto we were able to carry out a systematic comparison of
the δTot with the published total solubility (Hildebrand) parameters. We could
then see if it was reasonable to change any values using dipole moment and
refractive index data contained in those databases. The fundamental principle
of databases means that those databases also contain errors and conflicts.
Wherever possible we corrected those molecules where there was a large (>1)
difference in δTot, but used the principle of least change if DIPPR and Yaws
disagreed, and used the principle of common sense when a value in those
databases simply made no sense.
We have continued to work with Hiroshi to
challenge and revise the HSP database, especially when any fresh data appeared.
We continue to be hopeful that new
measurements of HSP (e.g. via IGC) will start to accumulate. See the next
paragraph for how you can help!
DIWF
The alternative to DIY is Do It With
Friends. The .hsd format is a simple text format that makes it very easy to
exchange HSP values. If members of the HSPiP user community email to Steve
their HSP values for chemicals not included in the official Hansen list then we
can start to share them amongst the community. Although each individual user
might be losing out by giving away some hard-won data, the community as a whole
will benefit. When different users come up with different values, we can choose
to quote both or launch a discussion to decide which is right.
Indeed, it might be time for those with
their private collections of HSP to open them up to the world-wide HSP
community. Of course they would lose some commercial/academic advantage by
revealing their values. But they would also gain by having those values
corroborated and/or refuted by values from other collections. By assembling one
large “official” HSP table, with differences resolved by expert assessment,
many of the glitches and problems in the literature and in our own practical
research enterprises would disappear. Will readers of this book take up the
challenge? We hope so!
E-Book contents | HSP User's Forum
