Utilities:
The
Utilities use as common:
Screen Capture
![]()
Table Capture
![]()
Clear Button ![]()
Calculation Button ![]()
Screen Capture
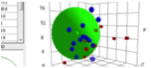
For a permanent record, click Capture Screen to Clipboard.
You can then paste the image into Word
etc. If you do Ctrl-Click you capture just the 3D
image. If you do Shift-Click you capture just the 3
2D images. The Tooltip on the button reminds you of which option is which.
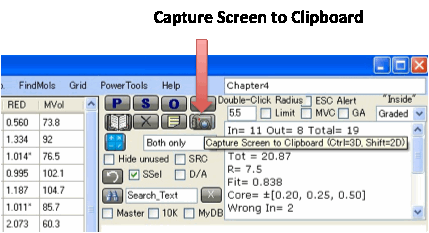
Table Capture
For a permanent text record of the
solvents, their RED numbers etc., click the Capture
to Table icon.
This pastes nicely into programs such as
Excel. For consistency within Excel, the “*” designations for Wrong In or Wrong
Out are removed – otherwise Excel interprets the RED column as a mixture of
numeric and text data. Note, too, that if you capture CAS Number data and place
it in Excel some of the numbers get automatically translated by Excel into
dates. This annoying feature of Excel has exasperated many people and can’t
easily be over-ridden.
If you just want a simple printout of
your table then select File Print Table.
If you want fancier printing then use
Capture to Table to place the data in Excel and use the many options there for
fancy print formatting.
Clear Button
To remove all the Inside/Outside solvent
designations in order to start afresh, press the X Clear button.
Calculation Button
![]() buttons
are located where you need enter calculation procedure.
buttons
are located where you need enter calculation procedure.
Utilities
located on Menu Bar
![]()
File: Open/Save file
Diff.: Diffusion (see Difusion Function)
Adh/Vis.: Adhesion and Viscosity calculations
Force Fit: If you know the Center of Sphere...
Teas: Make Teas Plot
HPLC: High Performance Liquid Chromatography
Simulation
IGC: Inverse Gas Chromatography Simulation
GC: Gas Chromatography Retention Index Simulation
Temp.: Temperature effect Calculator
Evap.: Evaporation Calculator
FindMols: Find (HSP range, Functional Group)
Molecular
Grid: Makeing Grid of Solvents Function
Power Tools: (see Power Tools)
Help: How To Use HSPiP
File:
When you File
Save
a dataset it's a good idea to name it as the polymer (or whatever) that is
being tested. If you need to remind yourself of the H,P,D,R data for that
polymer, simply load it and press Calculate.
Dist.:
Distance utility lets you
calculate the distance between HSP values.
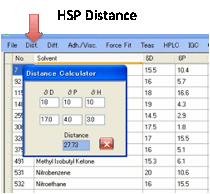
Each time you change one of the values,
the Distance is automatically updated.
This Distance is, of course, the square
root of 4(D1-D2)2 + (P1-P2)2 + (H1-H2)2.
Adh./Visc.:
The Adh./Visc. utility implements the Adhesion
and Viscosity calculations described in the eBook.
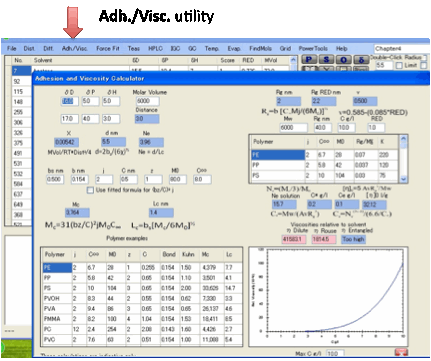
If you are interested in polymer/polymer
adhesion and polymer solution viscosity you will first need to read the section
in the eBook which describes what is going on. Tables of typical values for the
key parameters are included in the eBook. The calculated values are
automatically updated each time you change an input parameter.
Force Fit:
If you are convinced that you know the
true centre but want to know the Radius and quality of fit, click on the Force Fit option.
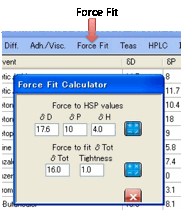
Simply enter your HSP values and click
the calculate button to the right of the HSP. (This option is also useful if
you want to enter some historical HSP fit for retrospective analysis). Or if
you are certain that you know δTot and want to constrain your fit, then
enter your δTot value plus an
estimate of “Tightness” of compliance to that δTot and click the calculate button to the
right of the δTot. The smaller the
value of Tightness, the more the fit is penalised for straying from your
requested δTot, but (in principle)
the worse the resulting fit. You can try a value of 0.5 (“Tight”) then 5
(“Loose”) to see what happens to the quality of your fit when forced to your δTot.
Teas plot:
For those who like triangular graphs
we’ve added the Teas plot, named after Jean Teas its
inventor.
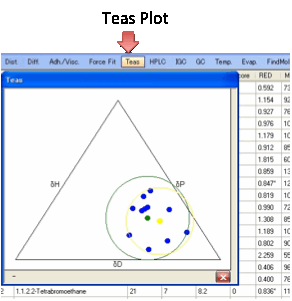
It simply plots δD/(δD+δP+δH) along one axis against δP/(δD+δP+δH) on the next and δH/(δD+δP+δH) on the third. Although this is a neat
way to condense 3D data into 2D, there is no scientific significance to the
plotted values! A perfect Sphere with all “good” inside and all “bad” outside
can look a muddle in the Teas plot. However, some people find it visually useful.
As a visual aid, the computed centre and radius are plotted using their own (δD+δP+δH) value along with the “bounding circle”
and its centre. Again these have no great scientific significance. When you
move your mouse over the Teas plot you get either the % δD, %δP, %δH or if you are near a solvent, the
actual δD, δP, δH and the solvent name.
HPLC:
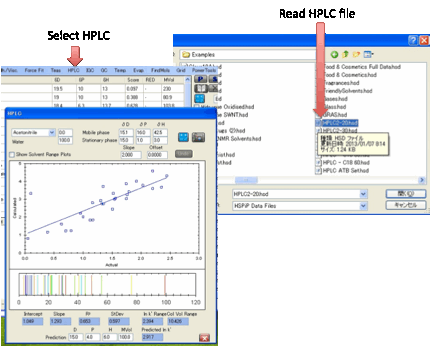
There is good scientific reason for
believing that there should be a linear relationship between ln k’ (the log of
the HPLC capacity factor) and the relative cohesive energy difference between
the analyte/stationary phase and analyte/mobile phase.
In other words, if you know the HSP of
your column and of the mobile phase then the retention time (as expressed via
ln k’) is easily calculable.
The problem is that you may not know the
HSP of your column and your estimate of the HSP of the solvent mix may also not
be too accurate. The solution to this problem is the HPLC modeller which lets
you adjust all the key parameters to see if the fit is meaningful or not.
If the data from your test materials is
meaningful then the modeller lets you predict the ln k’ of any other analyte.
It also lets you predict those two (usually competing) factors: speed of
separation and size of separation where you want a fast speed (short retention
times) combined with a large difference between analytes (long retention time
for the slower-moving analytes).
The first thing to do is to use the basic
Sphere form with your test materials. Instead of the normal 0-6 in the
Solubility column you enter the ln k’ values on your particular column. If you
then click the H button (for HPLC) you immediately get a form with a graph
showing your data fitted with whatever parameters happen to be active at the
time. Usually this will be a bad fit – as judged visually (large scatter) and
as judged by the R² value, the standard estimate of goodness of fit (a value of
1 is perfect, a value of <0.8 is really rather bad) and the Standard
Deviation (0 is perfect).
First check that you’ve selected the
correct solvent (Acetonitrile or Methanol) and the correct % solvent. This
gives a good estimate of the HSP of the mobile phase, but you can enter your
own estimate if you wish. Then it is recommended to set [15, 1, 3] as the value
for the stationary phase as this seems to be a good general starting point.
Now there are only 2 fitting parameters:
the Slope (C2) and Intercept (C1) of the formula:
ln k’ = C1 + C2 * MVol * (Distancema
– Distancesa)
where
the Distances are calculated using the normal Hansen equation and C1 contains
both the “column constant” and entropy terms and C2 is a combination of 1/RT
and a factor related to the number of theoretical plates in that particular
column.
To get
the best fit, click the Calculate button .Fitting routines can sometimes give
weird results – if they are too weird, click the Undo button and try changing
the values manually. Usually once you get close, the automatic fit becomes more
reliable.
If
just one of the data points is seriously out, it might well be that the
“official” HSP value is not good enough. Although many values are very well
verified, many of them are estimates that can (and this is openly acknowledged)
be out by 1 unit. If an adjustment by ~1 unit gives you a better fit then you
may well decide that you’ve found a better data set for that analyte and can
make it a permanent part of your overall data set.
Clearly
there is risk in adjusting data to give you the desired fit. At this stage of
the development of the HSP HPLC theory all we can say is that our experience
shows that the HPLC data seem to be more valid than some of the standard HSP
values so modest changes seem to be valid. As the theory and data sets evolve
we will be able to review this situation.
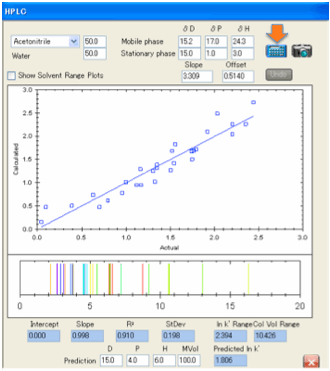
If the fit is generally poor then you
need to check whether you have entered the correct solvent blend and/or the
correct values for the stationary phase. Our
experience has been that most poor fits have been due to inputting errors on
our part (e.g. mixing up 60/40 with 40/60 blends) and not due to poor theory.
But once again you may find that HSP HPLC does not work for your analytes. We
would very much like to know counter examples and urge you to send us your .hsd
sets where the fits are bad.
With a good fit you can then explore what
would happen if you introduced a new analyte. Enter its HSP and MVol parameters
and you will get a good prediction of ln k’. Similarly, you can change column
parameters and/or solvent blend to see what the new predicted values would be –
maybe you can get faster separations with good resolution or maybe not. The
whole point of the modeller is to give you an intelligent method to improve
your HPLC separations.
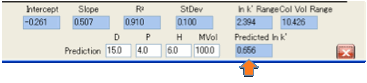
If you select Show Solvent Range Plots then for each analyte the calculated
values for the solvent blend ranging from 0 to 100% water are plotted in the
Y-axis with the X-axis value remaining the experimental value.
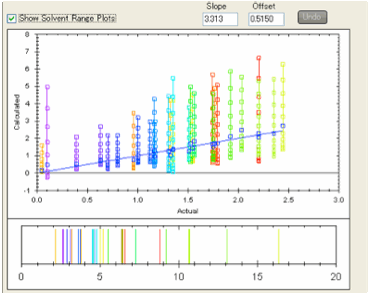
This gives you a quick graphical view of
how the relative retention times for the different analytes will change with
solvent blend. You can read off the values from the graph or you will find that
they are automatically placed onto the Clipboard in a format that is readily
pasted into spreadsheets such as Excel.
The simulated HPLC plot also gives you
visual indication of what happens if you change the solvent blend. By moving
your mouse over each peak you can find which analyte is which and also its
exact predicted column volume (where 1 column volume means 0 retention).
Finally, if you have confidence in this
methodology then you have a very quick way of getting good HSP estimates of new
analytes. From the ln k’ you can try out various HSP sets to see which gives a
sensible fit. You can use the DIY HSP to give you some estimates of what
sensible values (and MVol) might be. For example, if you find that [15, 3, 8]
and [15, 8, 3] both give a good fit, you can use your intuition (e.g. “this
molecule actually has an –OH group”) to help you decide which is the more
probable value. It is impossible to solve an equation
in 3 unknowns with 1 data point, so one HPLC run cannot give you unambiguous
numbers.
If you try three different solvent blends then you might get three data points
that are good enough to give you a precise unambiguous HSP set.
The point about multiple solvent sets is
important. It is very easy to get great correlations with one column, one
mobile phase and one data set. What we’ve found is that if you have a data set
containing multiple analytes measured at multiple solvent blends then it is
very, very hard to “fiddle” the results. That is why we have confidence in this
technique. Our very first fits (when we had little idea of what column HSP
should be) were fine till we challenged them with other solvent blends and they
were quickly shown to be useless. We then found fits that were robust across an
entire range of acetonitrile/water and methanol/water blends.
The glory of science is that hypotheses
are refutable. If we get enough data to suggest that this HPLC methodology is
invalid, we will happily remove it from the book and the software. Till then we
offer it as an exploratory tool in the knowledge that all our attempts to make
it fail have so far failed – in other words, in our non-expert hands, and with
the data sets we’ve encountered, the technique seems to work very well, just as
the theory predicts.
IGC
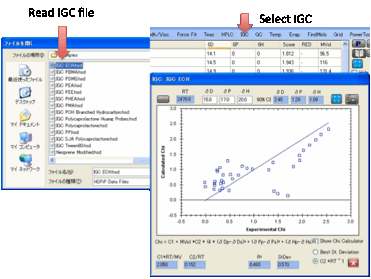
The basic theory of IGC is explained in
the eBook. “All” you have to do is create a .hsd file with Chi values in the
“Score” column, open IGC, and click the Calculate button. The best fit
HSP values are automatically produced for you and you can see the resulting
graph to show you how good the fit really is.
But of course things aren’t that simple.
First, many IGC experiments are run at high temperatures. For these you need to
click the ºC button, enter the IGC temperature and
click Calculate. This automatically calculates the HSP of the test solvents at
that temperature and also translates the best fit HSP on the IGC form (which is
the fit at the IGC temperature) down to room temperature. To do this the
thermal expansion coefficient of your material has to be estimated. If you
don’t know this experimentally then 0.0007 seems from the literature to be a
reasonable estimate for a wide variety of probes.
The second complication is that although
the calculated fit is guaranteed to be the mathematically best fit – only the
scientist can judge whether the fit really is any good. Two options have been
provided. The first attempts to minimise the standard deviation, the second
tries to get the data so that the C2 value is close to its theoretical value of
1/RT (and for convenience we calculate RT for you).
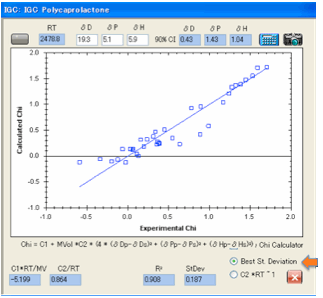
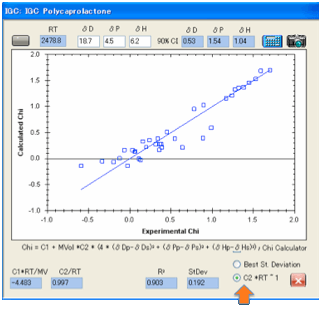
For many data sets we’ve tried the two
results are about the same (which is what they should be!) but if there is a
difference then you should recognise that many forms of error can creep into
experiments and you should use your best judgement as to which of the two makes
more sense.
There is a mathematical estimate of the
90% confidence levels, using Huang’s techniques described in the referenced
paper. This is a helpful guide, but the scientist’s judgement should also be
used. And the model makes this easy to do.
We strongly recommend that when you get
your calculated best fit that you should then manually vary the HSP parameters
and see what happens to the fit. If you have an optimum set of probe solvents
(ones that fully encompass the HSP of the test material) then any change of
parameters should make the fit significantly worse. But if your probes don’t
cover a full range (and this is often the case for dD) then relatively large
changes in a parameter will make relatively small changes to the quality of the
fit. If you find this situation then you have two choices. First, to run more
(or, rather, different) solvent probes to better cover the HSP range. Second,
to use your judgement and, maybe, the DIY section to make a sensible decision
on the optimum values. For example, if the “best” fit for a material which is
largely aliphatic comes out with δD=21 and you find that the quality of the
fit is not much different with a more expected δD=17 then go for the 17 rather than the
21.
So far we’ve assumed that you have a set
of Chi values for the solvents in relation to the stationary phase. However,
it’s really rather tricky to calculate these from the prime data, Vg, the
relative retention volume. You need Antoine constants and B12 (Second
Virial Coefficient) values for each of the solvents. If you have all these then
that’s not a problem. But for those who don’t we’ve added a Chi calculator that
opens up when you click Show Chi
Calculator.
We’ve provided a selection of typical IGC
solvents along with their Antoine constants and Critical Parameters. From the
Critical Parameters we can estimate B12 by one of two methods from the
literature. The first one uses Tc and Pc, the second uses Tc and Vc. One tends
to underestimate B12, the second tends to overestimate, but this is
only a general trend and there are many exceptions. The point of providing
these two methods is that you can check for the effect of the estimated B12
on your final HSP calculations. If B12 values are critical for the
HSP then you need to find better estimates. If, as is often the case, the B12
values make only a modest difference, then you can relax a little. The B12
value is entered in the table, but you have to scroll over to the right to find
it.
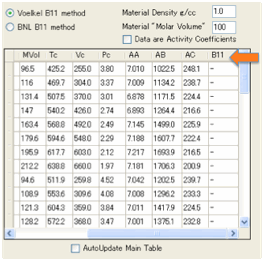
The assumption is that when you enter
your Vg in the Chi calculator that the Chi value in the main .hsd table should
be automatically updated. However, if you just want to play with Vg-to-Chi
without changing the Chi values in the main table, de-select Auto Update Main Table.
Some literature provide IGC outputs as
Activity Coefficients rather than raw Vg or calculated Chi values. To use such
data, make sure the Show Chi Calculator option is checked, then enter the Molar
Volume and Density for the stationary phase and select Data are Activity Coefficients. As long as your
solvents have been selected from the master list, their names will match those
in the master list and the solvent density is automatically calculated from the
molar volume and molecular weight in that list. If not, you need to enter the
solvent density manually into the only convenient place for it – the RED number
column. Any entry in this column (other than the default “-“) will override the
calculation from the master list.
GC:
It would be very useful if we could
predict in advance the exact retention time of a chemical on a GC column. But
of course this is not directly possible as retention time depends on numerous
specific variables in the GC analysis: column type, column dimensions,
temperatures, flow rates etc.
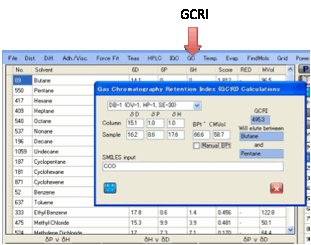
A useful substitute is the GCRI. We know
that the retention time of straight chain alkanes form an orderly progression
from CH4 to C2H6 to C3H8 … And if we give each of these alkanes a retention
index = 100 x number-of-carbon-atoms we can say, for example, that if a
chemical elutes at precisely the same time as heptane then its GCRI is 700.
If the chemical elutes somewhere between
hexane and heptane then, by definition, the GCRI is somewhere between 600 and
700. Kovats proposed a formula for calculating the GCRI. Suppose the lower
n-alkane elutes at time A, the higher (n+1)-alkane elutes at time B and the
chemical elutes at a time X between A and B then:
GCRI = 100 * ( log(X) - log(A))/(log(B) -
log(A)) + n * 100
If, in the example above, hexane eluted
at 60 seconds, heptane at 80 seconds and the chemical at 70 seconds then GCRI =
100 * (1.845 - 1.778)/( 1.903 - 1.778) + 600 = 654
If we can predict GCRI from the chemical
structure then we can provide an accurate estimate of the retention time in a
specific column provided we know the retention times of the alkane series.
For the HSPiP predictor we need to know
three things:
The HSP of the column
(typically [15, 1, 1]
The HSP of the molecule
The Boiling Point of the
molecule
The properties of the chemical are either
known (in which case you can simply enter them into the boxes) or can be
estimated using the Y-MB technique from the SMILES of the molecule.
Of course if you are combining predicted
HSP and predicted Boiling Point with a GCRI estimator which uses a fitted
relationship based on differences in δD, δP and δH, it is unlikely that the predicted GCRI
will be highly accurate. But in our experience it is a very good starting point
for thinking through GC conditions that would allow good analysis of your
molecule. You set up the column so that the alkanes either side of the
predicted GCRI (which are named in the output) are separated quickly and
efficiently, then try your molecule. There is a good chance that you will be in
the right range!
Temp.:
Temperature
effects
HSP change with temperature according to
the formulae:
d/dT δD = -1.25á δD
d/dT δP = - á δP/2
d/dT δH = -(1.22 x 10-3 + á/2)δH
Unfortunately, á the thermal expansion coefficient is not
a constant and has to be calculated for each temperature. The “Te-a, Te-c…”
values in HSPiP Solvent Data are the parameters used to calculate the thermal
expansion coefficient at different temperatures. Even more unfortunately, for
many chemicals we don’t have data for á or its temperature dependence. So it’s
not easy to get accurate values at different temperatures. However, we do our
best. If you click the Temperature menu option you can
select a temperature and a default value for á for those cases where it is unknown or
outside its specified thermal range. Clicking Calculate shows the change in
HSP.
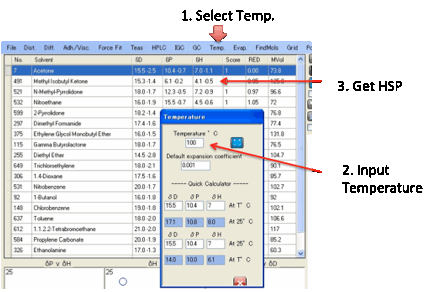
You can then do Sphere fitting with these
revised values if your experiments were done at your chosen temperature. The
standard temperature is assumed to be 25ºC so if you enter this value you just
get the plain value. As these calculations aren’t amazingly accurate we round
the temperature to the nearest integer value.
Evap.:
The Evaporation utility gives you the
following useful numbers:
Vapour Pressure at the
given temperature
Relative Evaporation
Rate v nBuAcetate = 100
Relative Evaporation
Rate v Diethyl Ether = 1
Absolute Evaporation
Rate in µm/hr/cm2
Absolute Evaporation
Rate in mg/hr/cm2
The inputs are the Molar Volume and MWt
of your solvent, the temperature, the wind speed (which affects absolute, not
relative rates) and the Antoine Constants, AA, AB, AC (in units that give the
vapour pressure in mm/Hg). The Antoine Constants can be obtained from standard
databases such as Yaws or can be estimated by Y-MB using the SMILES input. The
constants for the two reference solvents are shown for convenience.
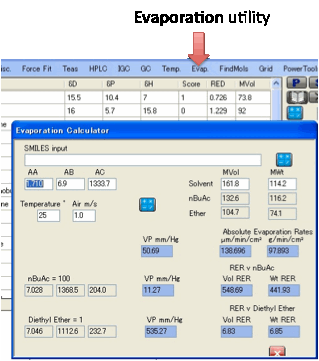
The outputs show the vapour pressures of
your solvent and the two reference solvents, plus the RER with respect to those
reference solvents. The values for Ether are confusing because they are
actually the relative time taken to evaporate
rather than the relative evaporation rate. So a higher RER for nBuAc means a lower RER for Ether and vice-versa.
You will notice that the calculated RER
can differ from those in various databases (including the RER in the Solvent
Optimizer). It has to be said that RER isn’t the most precise constant in any
database – and if you check on various databases you often see different
numbers. Real evaporation rates depend, for example, on whether the evaporating
solvent cools itself which, in turn depends on heat transfer coefficients. A
layer of solvent evaporating on top of some insulating layer (e.g. a foam) will
evaporate more slowly than one on a metal tray. The difference is larger for
faster-evaporating solvents so the measured RER of a volatile solvent may be
lower (depending on thermal conductivity, air speed etc.) than predicted from
the vapour pressure at a given temperature.
There is a common error in thinking that
RER depends solely on relative vapour pressure. But vapour pressures work in
moles so molecular weights and molar volumes are significant when you are
comparing weight-based or volume-based RERs.
FindMols:
The large HSPiP database containing
predicted values gives you a chance to hunt for alternative molecules. In FindMols you can search in two ways.
•
Find molecules that fall within a certain range of δD, δP, δH (or δΗdo and δHac if you like split H-bonds) or BPt.
Hopefully the way that this works is obvious.
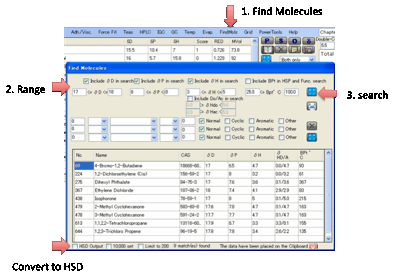
•
Find molecules containing certain (combinations of)
functionality. This is a bit more complex. Suppose you want to find Cyclic
molecules containing exactly 2 C=O groups. You select 1 < C=O < 3 and
check just the Cyclic box. If you wanted to refine the search to include
molecules with 1 or 2 Cyclics with C=O and also with a single –NH2 group, you
would change the first line to 1 = C=O < 3 and add a second line, 1 = NH2
< 2 with Cyclic also checked. If you select the BPt option above then you
further restrict the values that appear.
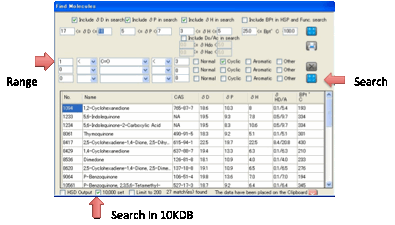
In both cases the list of predicted
values is shown in a table (for “official” solvents the HSP values are official
and many of the values are real data).
The list you create is automatically placed on the Clipboard in a format
that is easy to paste into Excel. If you want the list as a .hsd file that’s
then automatically imported into HSPiP, check the HSD Output option. You can choose to search in the
standard HSP list or by checking the 10K option, the entire
database. You can also choose whether to limit your search to a maximum of 200
in case your criteria are too broad.
Grids:
An alternative way to create a set of
solvents for the Sphere method is to use mixtures of pairs of solvents. The Grid option allows you to choose pairs of solvents, combined in
a chosen number of Grid Steps and by clicking the Create button automatically generates the combinations and shows
them as a plot in 3D graph.
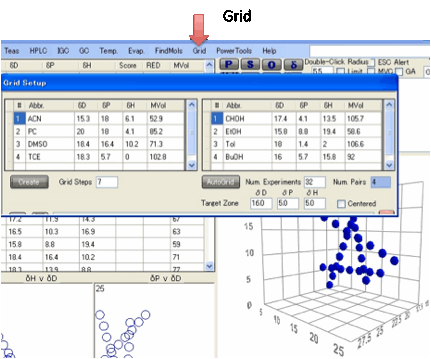
Rather than use a full solvent name it is
best to use an Abbreviated name. The HSP values can be put
into the rows via the normal Ctrl-D from a master list and Ctrl-P into the
appropriate rows.
The solvent choices can be loaded and
saved as .grd files.
As a further option an AutoGrid can be created that is targeted to a
particular area specified by the Target
Zone
inputs. By choosing Num. Experiments you intend to make and
the Grid Steps this automatically works out the Num.
Pairs
to be created. The algorithm takes all the solvents currently in the Solvent
Optimizer (as a convenient source of practical solvents) and examines each pair
of solvents to see if they are useful for the Grid – i.e. they produce a scan
line that crosses the target zone. If the Centered option is chosen then
there is a bias towards scan lines that start and end on either side of the
sphere.
It has proven very hard to decide on an
optimum AutoGrid algorithm. But
what is clear is that more important than the algorithm is the choice of
solvents to be used! In other words, please put a lot of thought into the range
of solvents within the Solvent Optimizer. It’s a good idea to carefully create
a special Grid set which covers a good range of Grid space without too many
duplicates. So there is no point in having both hexane and heptane in your
solvent list because if a hexane:propylene carbonate scan line is good then
it’s likely that the AutoGrid would also find that a heptane:propylene
carbonate scan line would also be good – whereas it adds no real new data.
Please note that the grid is merely a way
to do the boring work of setting up a reasonable solvent range – but users
don’t have to use it blindly.
The user can easily
delete points they don’t like and, if they want, add some pure solvents in key
points. This is done in the main table, as usual.
The user has to check
for miscibility and take into account MVol effects with, e.g. swelling.
The user can move the
ranges based on their intuitions of where the test material will be.
It can be used to refine
the radius if a solvent close to the middle is chosen as one of each pair.
