Molecular Orbital & Properties: TSL: The level of ab-initio calculation
For guessing a reactant ratio by calculation Which Level Ab-Initio Calculation?
I am an experimental researcher and honestly it is a mystery for such a person as how much the calculation level of un-experiential molecular orbital calculation should be set. It being the same as a experiment and applying a trial-and-error method, please think the account of whole circumstances which has verified the calculation level, and read. (About the contents, it cannot have responsibility.)
By the difference in a solvent, the difference in an initiator, and the difference among polymerization conditions, a reference value is also large and, as for the reference value of a reactant ratio, change stripes use this value this time.
| r1 |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
MalR |
AMR |
VFR |
| St |
1.00 |
0.46 |
0.33 |
0.09 |
0.02 |
0.10 |
0.10 |
0.18 |
|
| MMA |
0.52 |
1.00 |
0.15 |
0.25 |
0.07 |
0.02 |
0.02 |
0.34 |
|
| AN |
0.40 |
1.20 |
1.00 |
0.60 |
0.04 |
0.06 |
0.00 |
0.95 |
0.005-1 |
| VDC |
2.00 |
2.75 |
0.80 |
1.00 |
0.30 |
0.05 |
0.00 |
0.65 |
0.16 |
| VC |
17.00 |
16.10 |
2.70 |
3.20 |
1.00 |
0.23 |
0.01 |
4.40 |
0.11 |
| Vac |
55.00 |
20.00 |
4.05 |
6.70 |
1.68 |
1.00 |
0.00 |
9.00 |
0.25 |
| Mal |
0.01 |
3.50 |
6.00 |
9.00 |
0.30 |
0.06 |
1.00 |
2.80 |
|
| AM |
0.75 |
1.69 |
1.40 |
0.85 |
0.12 |
0.10 |
0.02 |
1.00 |
0.01 |
| VF |
|
|
24-44 |
6.00 |
11.60 |
3.50 |
|
43.00 |
1.00 |
Calculation was started by HF/6-31G**, without understanding anything that he wants to carry out calculation which reproduces such a reactant ratio. Styrene and acrylonitrile are that calculation suits neither a radical nor a monomer at all that I understand immediately first.
| HF6-31G** |
StR |
ANR |
VacR |
| St |
1.00 |
0.00 |
1.22E-14 |
| MMA |
29694586.87 |
28.19 |
0.03 |
| AN |
93538.02 |
1.00 |
0.00 |
| VDC |
13852426.34 |
62.58 |
0.02 |
| VC |
95205378.32 |
153.23 |
0.16 |
| Vac |
1383386471.98 |
615.97 |
1.00 |
| Mal |
4896268.47 |
20626.79 |
0.01 |
| AM |
14419276.22 |
31.65 |
0.07 |
| VF |
2502519489.70 |
1841.68 |
3.23 |
An order is different or it is not the talk of a thing level. However, if the two are removed, it can be said that the reaction of the radical (VacR) of vinyl acetate suits very well to monomers other than St and AN. Then, if the level of calculation was raised by the sadness which nothing knows, when it might improve, it calculated about AN and St by HF/6-311G**.
No-
Time has not improved at all, although very started. then, in the system that electron correlation will involve if a reference book is read by the HF method, if incalculable, it is. And it is "Carry out MP2 calculation."
Then, although it is a reason for having tried at MP2 calculation, in St-St, it is too large and calculation is impossible. The relation between a memory and a disk is not known well somehow. Seemingly a memory is not an excuse, either, if a memory is increased, although there are 2GB and a 100-G disk, and it is not an excuse, either, if Maxdisk is increased. A way was not found well in any way, but MP2/6-31G** gave up, and, moreover, was calculated only within small monomers, such as acrylonitrile and vinyl chloride, by MP2/6-31G.
Also NO-
Unless MP2 calculation combines with a basis function big as it is, doesn't an appropriate answer come out of it? Starting in time , the result brought the result that he had HF and no great difference.
If a reference book is taken out and read again, when you calculate a big molecule, calculate structure with a low basis function and calculate only the energy with a high basis function. It is.
MP2/6-31G**//RHF/6-31G**
Seemingly, it will be written as. it is surely this which is not obtained. Even if it considers, the method of submit of such a type of job is not known again. Useless, although the homepage of whether there is any sample and a teacher is visited. I did his best in the try and error again. How should especially a setup of a memory and a disk be done? And after calculation is completed somehow, there is three negative vibration!!!
It seems that a negative vibration appears a lot on MP2 level also according to the transition state carried out perfectly in HF / 6-31G** level. Although FORCE calculation is carried out here in order to calculate a frequency factor in quest of an entropy clause, when consulting with a teacher, it has been said that such a way is a way about which it argues only by dE, and it all must be done by MP2/6-31G** if you want to calculate it to vibration.
Well Everything going wrong
The re-calculation was done by B3LYP/6-31G** as having nothing to lose. It is because "The transition state of B3LYP was not so good although B3LYP is good for calculation of a stable compound" was said by the teacher on whom a lecture was given by the CAC forum when it said why this was not used from the beginning. Although its calculation also differed in the system, it actually had the example which does not suit an experimental value at all by B3LYP.
| B3LYP |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
malR |
AMR |
VFR |
| St |
1.00 |
0.34 |
0.20 |
1.10 |
0.16 |
13.13 |
0.00 |
0.59 |
0.03 |
| MMA |
0.30 |
1.00 |
0.70 |
0.13 |
0.07 |
0.05 |
0.00 |
0.61 |
0.00 |
| AN |
0.05 |
0.09 |
1.00 |
0.11 |
0.03 |
0.01 |
0.04 |
2.13 |
0.00 |
| VDC |
1.22 |
1.20 |
2.42 |
1.00 |
0.24 |
0.12 |
0.02 |
2.26 |
0.02 |
| VC |
8.98 |
7.16 |
9.18 |
1.66 |
1.00 |
1.01 |
0.06 |
16.86 |
0.17 |
| Vac |
20.36 |
5.69 |
5.86 |
1.61 |
2.66 |
1.00 |
0.02 |
11.42 |
0.29 |
| Mal |
0.03 |
0.38 |
229.14 |
4.89 |
0.08 |
0.00 |
1.00 |
2.87 |
0.00 |
| AM |
0.11 |
0.30 |
0.80 |
0.11 |
0.10 |
0.11 |
0.01 |
1.00 |
0.01 |
| VF |
121.45 |
14.60 |
34.43 |
2.94 |
6.33 |
7.71 |
0.06 |
25.80 |
1.00 |
In this calculation, St and AN are calculable somehow. The thing and yellow in which pink is contained within 3 times (1/3) have an unknown experimental value. I think that it can calculate fairly well. Compare the result of this, and HF/6-31G**.
| HF6-31G** |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
malR |
AMR |
VFR |
| St |
|
|
|
|
|
|
|
|
|
| MMA |
|
1.00 |
|
0.10 |
0.16 |
0.03 |
0.00 |
0.63 |
0.01 |
| AN |
|
|
|
|
|
|
|
|
|
| VDC |
|
0.57 |
|
1.00 |
0.27 |
0.02 |
0.03 |
0.76 |
0.01 |
| VC |
|
1.48 |
|
0.72 |
1.00 |
0.16 |
0.05 |
5.55 |
0.08 |
| Vac |
|
6.40 |
|
2.19 |
6.38 |
1.00 |
0.24 |
17.62 |
0.53 |
| Mal |
|
0.53 |
|
11.48 |
0.70 |
0.01 |
1.00 |
1.37 |
0.01 |
| AM |
|
0.42 |
|
0.12 |
0.20 |
0.07 |
0.00 |
1.00 |
0.01 |
| VF |
|
10.36 |
|
2.48 |
13.93 |
3.23 |
0.26 |
29.79 |
1.00 |
It is the calculation result of HF/6-31G**, and the light blue was attached to the thing near an experimental value rather than B3LYP/6-31G**. In a monomer, styrene, acrylonitrile, MMA, AM, etc. are useless by the HF method. That is, it is useless [ the HF method ] to a compound which has double bond next to double bond. It seems that maleic anhydride does not almost have a difference. This neighborhood is wonderful. Conversely, vinyl chloride, a fluoridation vinyl monomer, etc. have achieved results with the more sufficient HF method. It is the result of saying that it hardly changes about a VDC monomer.
Calculating all by B3LYP also gives up, and it just called the halogenated compound calculated by HF
It seems that an MMA monomer also suits better than B3LYP if it will calculate on a HF/6-311++G** level supposing a margin is in a machine, a memory, and time.
| HF6-311++ |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
malR |
AMR |
VFR |
| St |
|
|
|
|
|
|
|
|
|
| MMA |
|
1.00 |
|
0.26 |
0.12 |
0.026 |
0 |
0.504 |
0.005 |
| AN |
|
|
|
|
|
|
|
|
|
| VDC |
|
0.16 |
|
1.00 |
0.14 |
0.011 |
0.01 |
0.214 |
0.0061 |
| VC |
|
0.85 |
|
1.72 |
1.00 |
0.136 |
0.03 |
3.392 |
0.0956 |
| Vac |
|
5.69 |
|
7.90 |
7.45 |
1.00 |
0.18 |
14.86 |
0.5767 |
| Mal |
|
0.42 |
|
19.07 |
0.455 |
0.007 |
1.00 |
0.844 |
0.0057 |
| AM |
|
0.50 |
|
0.38 |
0.202 |
0.058 |
0 |
1.00 |
0.0089 |
| VF |
|
8.87 |
|
8.43 |
15.51 |
2.32 |
0.28 |
22.6 |
1.00 |
Since an overall tendency does not simply change, it is just going to divide judgment whether it does so far. Yellow is attached to the thing near an experimental value rather than B3LYP/6-31G** what has unknown pink and an unknown experimental value.
If calculation of such a level is early and will be one day slow, it also has one month also of these things. It just said with three to four days that it averaged. (Being about Mac G4 1GHz.) at Windows, it has about 1.5 times earlier sensibility by the same Hz. although it was probably various whether this is regarded as early or it was concluded that it is late, the activation energy of PM3 of MOPAC was guessed by the neural network -- as -- this The neural network which can reproduce the calculation result of B3LYP Build.
Ab Initio calculation, even easy Transition State Search takes 1 day long. If complicated TS Search takes one month or several month. It takes 3-4 days on the average (with Mac G4 1GHz) . Windows machine is about 1.5 times faster compare to MAC by the same Hz.
BB3LYP level calculation with neural network
although it was probably various whether this is regarded as early or it was concluded that it is late, the activation energy of PM3 of MOPAC was guessed by the neural network -- as -- this The neural network who can reproduce the calculation result of B3LYP Build.
If it is made, an answer should come out in a moment.
Although it is activation energy first, if a transition state can be found, it is in a calculation result.
Zero-point correction= 0.147422 (Hartree/Particle) |
The value of enthalpy and entropy is extracted. It extracts similarly from the radical and monomer of a raw material, and the difference before and after a reaction is taken. And the following tables are made.
name |
dE |
RaSOMO |
RaLUMO |
RaHCharge |
MonHOMO |
MonLUMO |
MonTCharge |
AA-AN |
6288.6127 |
-10.118 |
-0.814 |
-0.1489 |
-10.886 |
-0.187 |
-0.099 |
AMD-AMD |
6731.0468 |
-10.024 |
-0.48 |
-0.1804 |
-10.925 |
0.143 |
-0.0608 |
AM-VC |
8296.1964 |
-10.017 |
-0.711 |
-0.1578 |
-9.837 |
0.704 |
-0.1621 |
AMD-AllC |
8110.4368 |
-10.024 |
-0.48 |
-0.1804 |
-10.269 |
0.621 |
-0.1398 |
AA-Vac |
6889.1934 |
-10.118 |
-0.814 |
-0.1489 |
-10.083 |
0.687 |
-0.2102 |
VDC-St |
6283.5922 |
-8.803 |
0.16 |
-0.3909 |
-9.13 |
-0.122 |
-0.1581 |
AA-Acro |
4353.8267 |
-10.118 |
-0.814 |
-0.1489 |
-10.681 |
-0.098 |
-0.0493 |
Such a thing is prepared by 247 groups in which the transition state can be found in B3LYP/6-31G**. The first column is a name and, next, it is the amount of change of enthalpy (activation energy). And six subsequent columns are the explanation factors explaining activation energy, and although this is bean paste, the physical-properties value of the time of MOPAC PM3, the radical similarly calculated by MOPAC, and a monomer is contained in the value here. That is, the calculated value of B3LYP is guessed from the calculated value of MOPAC. If you want to predict the activation energy to a new monomer and a radical new transition state, it can be made to perform prediction of a B3LYP level only by calculating a raw material by MOPAC.
The item to input is completely the same as the time of doing by six kinds of radical SOMO, LUMO, the charge of a radical head, HOMO of a monomer and LUMO, and a monomer tail charge. The middle layer neuron's number and bias are also the same as a front, and are made to learn.
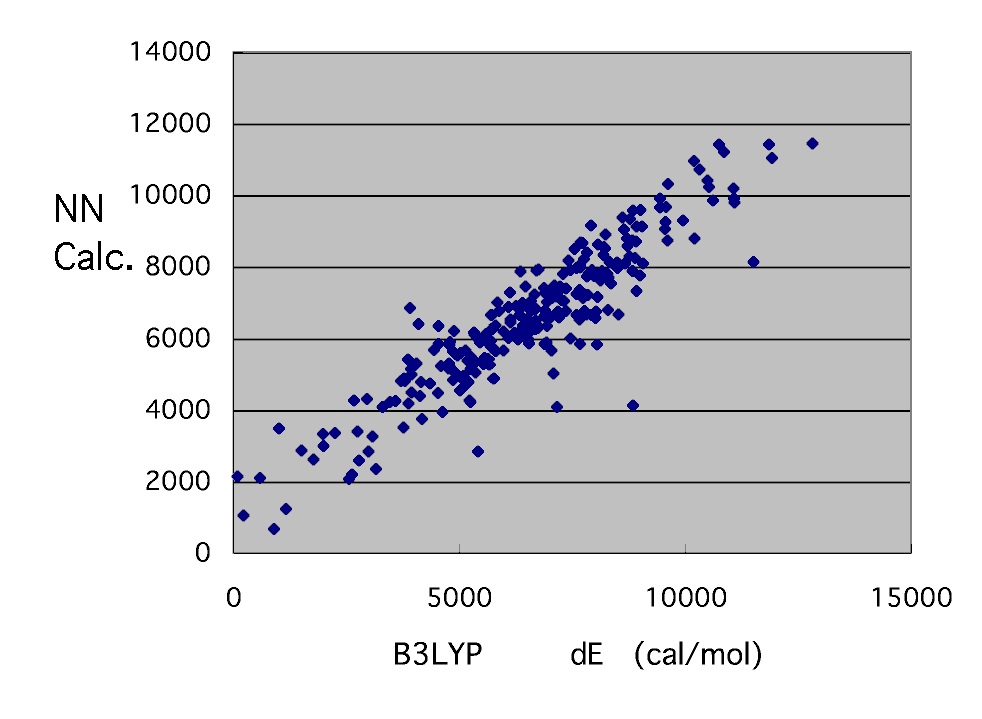
Although there are some clearly strange points, unlike MOPAC, it cannot re-calculate easily and cannot correct. It will remove for the time being and will check slowly later. It seems that a strange thing does not have even a system.
And it is in the last target.
Such a result was obtained. Although dispersion is large per [ with small dE ] two to 4 kcal/mol, it can be said that the result of B3LYP can be reproduced fairly good.
In the next The guess of a frequency factor A .
Frequency factor
The book with which how to ask for a frequency factor is explained is only Mr. Fujimoto's book , as far as I get to know. What is it why? Since it is hard to come out of calculation accuracy to one and hard to carry out track record contrast, will it be omitted?
In the formula, the frequency factor of two molecular reactions is written down as follows.
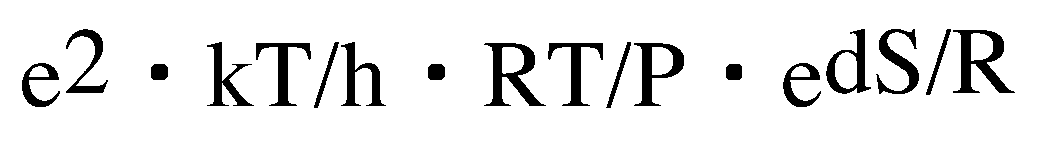
k: Boltzmann's constant
h: Planck's constant
. And the change clause of the entropy which asked for the point is put in and calculated to the place of dS. Temperature is made into 60 degrees C. The frequency factor of 247 transition states calculated by B3LYP/6-31G** is calculated. Since the value is small, the 17th power of 10 is hung.
Now, consider guessing this frequency factor by a neural network.
name |
A8 |
RaSOMO |
RaLUMO |
RaHCharge |
MonHOMO |
MonLUMO |
MonTCharge |
MMA-Mal |
14.528782 |
-9.566 |
-0.598 |
-0.1579 |
-11.708 |
-1.548 |
-0.1308 |
Vac-Amb |
724.54424 |
-8.785 |
0.553 |
-0.1144 |
-11.067 |
-0.078 |
-0.059 |
Vac-AA |
592.13286 |
-8.785 |
0.553 |
-0.1144 |
-11.147 |
-0.17 |
-0.0547 |
AA-Vac |
290.93853 |
-10.118 |
-0.814 |
-0.1489 |
-10.083 |
0.687 |
-0.2102 |
St-VF |
439.34772 |
-8.565 |
-0.225 |
-0.1462 |
-10.597 |
0.714 |
-0.2065 |
AMD-Mal |
192.37047 |
-10.024 |
-0.48 |
-0.1804 |
-11.708 |
-1.548 |
-0.1308 |
VC-AMD |
390.53817 |
-8.928 |
0.56 |
-0.3047 |
-10.925 |
0.143 |
-0.0608 |
AM-AA |
944.12965 |
-10.017 |
-0.711 |
-0.1578 |
-11.147 |
-0.17 |
-0.0547 |
Use as an explanation factor six kinds of radical SOMO calculated by MOPAC as well as the time of activation energy, LUMO, the charge of a radical head, HOMO of a monomer and LUMO, and a monomer tail charge, and a frequency factor is guessed by a neural network.
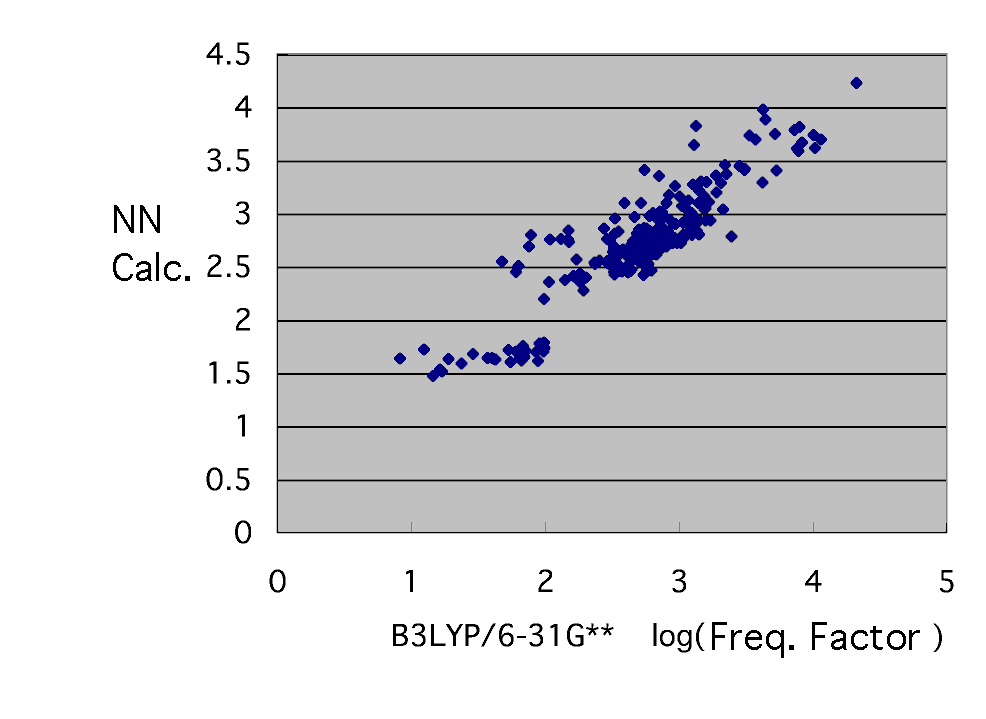
If Log is taken for a vertical axis and a horizontal axis and a graph is written, it is as mentioned above. It seldom converges. This has suggested that a frequency factor cannot be explained in six kinds of radical SOMO, LUMO, the charge of a radical head, HOMO of a monomer and LUMO, and a monomer tail charge. If it does so, it will consider either that what kind of item has next affected the frequency factor, or it is not needed among the above-mentioned six. The actual condition of a frequency factor thinks that change of entropy, i.e., a frequency, will involve, and considers the explanation factor which is likely to change a frequency. (Although it is XXXX which I considered, it does not dare write it here.) Please think also by yourself.
The neural network who guesses the frequency factor which he finally got has brought the following results by eight explanation factors and the middle layer neurons 7.
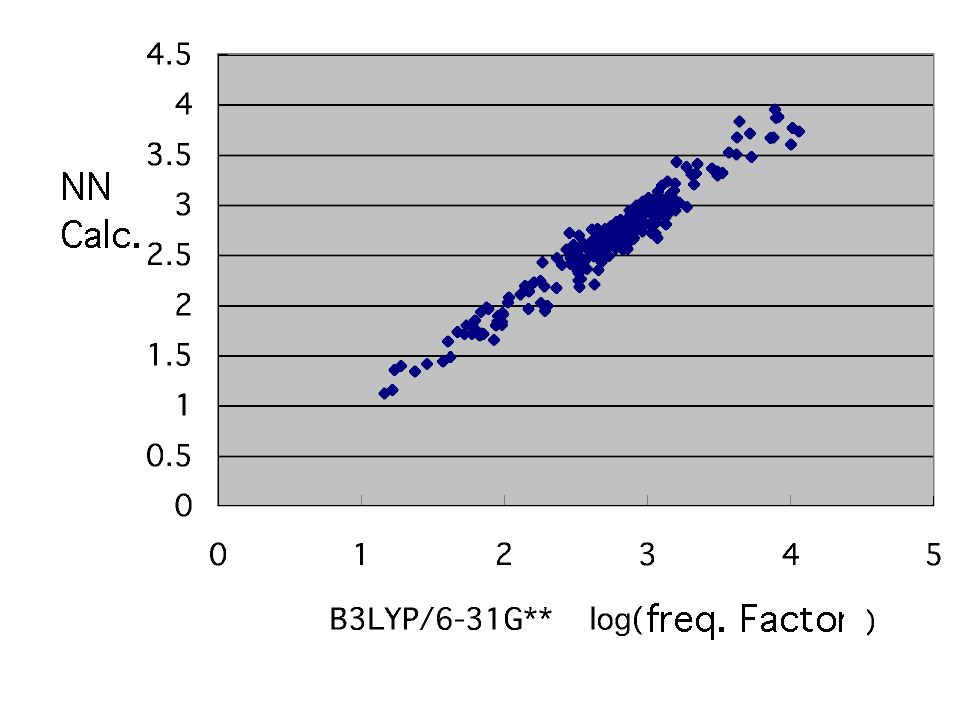
Thus, the frequency factor has been guessed only from the molecule information on a radical and a monomer. It says [ that a frequency factor and activation energy can be found ]. Reaction rate constant kij
kij=Aij*e(-dE/RT)
It is found at arbitrary temperature.
Reactivity constant kij
Reaction rate constant kij
kij=Aij*e(-dE/RT)
-- it is found at arbitrary temperature.
Since a frequency factor cannot calculate well in calculation by MOPAC, it is the ratio of a reaction rate constant. It came out and argued using certain r1r2. In Ab Initio calculation, since it was calculable to the frequency factor, it asked for the reaction rate constant. Since it compared how much this suits reality, the data of a growth reaction constant (kp) was collected. Mr. Imoto's textbook -- Professor Bagdasarian's data -- polymer -- as radical -- VAc, AM, AN, MMA, 2-VP, and MAN, St, Bd and I And VAc, VC, VDC, AM, AN, MMA, 2-VP, MAN, St, BD and I, chloroprene, and the 60-degree C growth reaction constant of Mal are recorded as a monomer. Other than this, the growth reaction constant in the case of homopolymerization (kp) appears also in handbooks, such as a polymer handbook.
however, this -- a trouble -- for example
In the case of acrylamide
| Solvent | Temperature | kp(l/s mol) |
| Water | 30 | 79000 |
| Dimethyl sulfoxide | 30 | 3700 |
| Dimethyl sulfoxide | 40 | 5000 |
In the case of acrylic acid
| Solvent | Temperature | kp(l/s mol) |
| Water | 30 | 31900 |
| Formamide | 30 | 5700 |
| Dimethyl sulfoxide | 30 | 500 |
It is difficult to sense, to come out and such to extract a value to one very much.
moreover, the case of Ab Initio calculation -- for example
| Bagdasarian | B3LYP/6-31G** | HF/6-31G** | HF/6-311++G** | |
| Vinyl acetate | 1700 | 830229 | 219 | 15.4 |
| Vinyl chloride | 1700 | 479957 | 610 | 109.7 |
| Methyl acrylate | 1260 | 242618 | 13.8 | 0.86 |
| Methyl methacrylate | 513 | 248 | 0.0199 | 0.00496 |
A result will change a lot with the calculation method and accuracy.
Compare with Professor Bagdasarian's data first here.
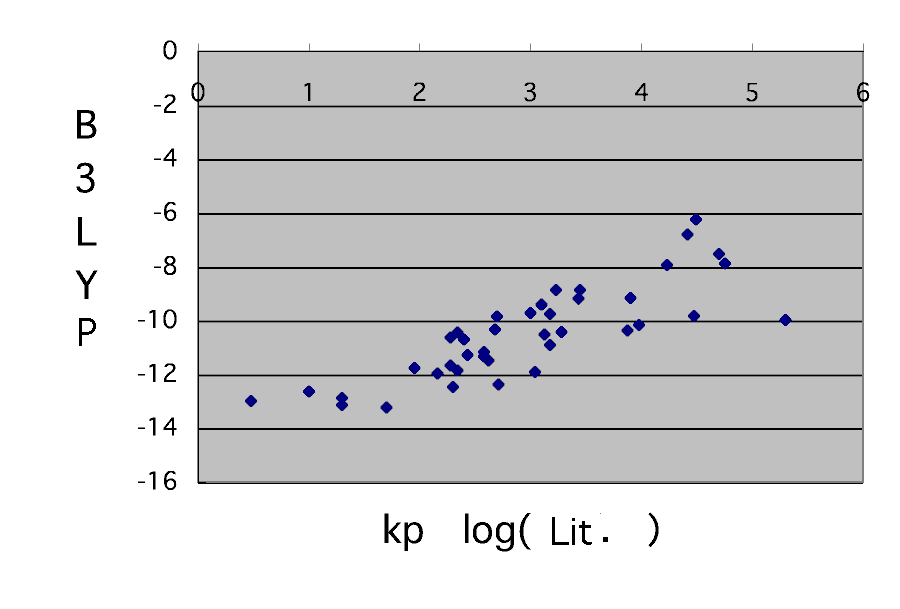
although it is unavoidable that an absolute value does not suit it is saying that a reaction rate constant is computable to such a grade with molecular orbital calculation. It is the place where it is difficult to be generally unable to declare this that the accuracy of a calculation result is low. An about double figures experimental value will also move also by the case of the upper acrylic acid with a solvent.
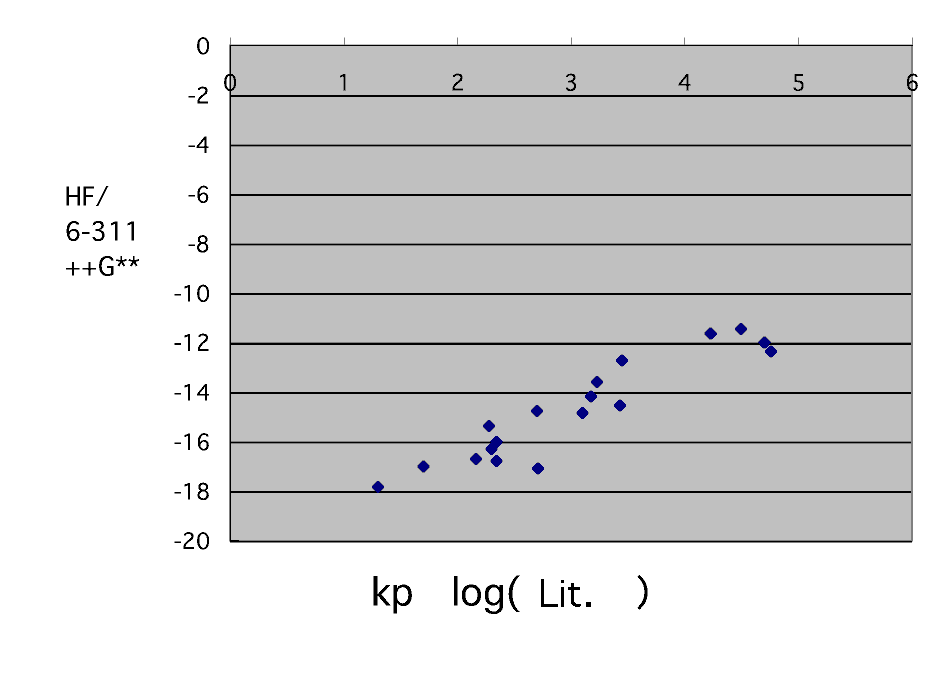
It seems that it will ride on a straight line still better if it calculates on a HF/6-311++G** level. However, styrene, 2-vinyl pyridine, acrylonitrile, and methacrylonitrile are incalculable.
I want to know the calculation result in B3LYP/6-311++G** . (Would you do someone ?)
It seems that the reaction rate constant obtained by molecular orbital calculation is systematically with error with an actual reaction rate constant with the permittivity (is it 1 ?) in the inside of a vacuum. there is also no way referred to as calculating taking permittivity into molecular orbital calculation -- coming out -- although it is not, it does not do this time.
If it says how many the reactant ratios r1 of an experimental value can reappear when it calculates on B3LYP / 6-31G** level,
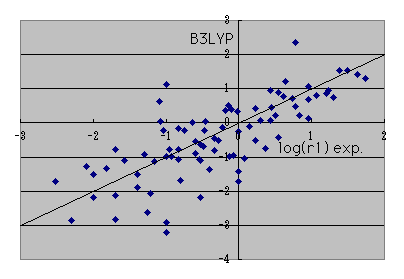
It is such a thing in a log plot. At an actual reaction, since r1 changes a lot only by a solvent changing, I think that it is such a thing in accuracy.
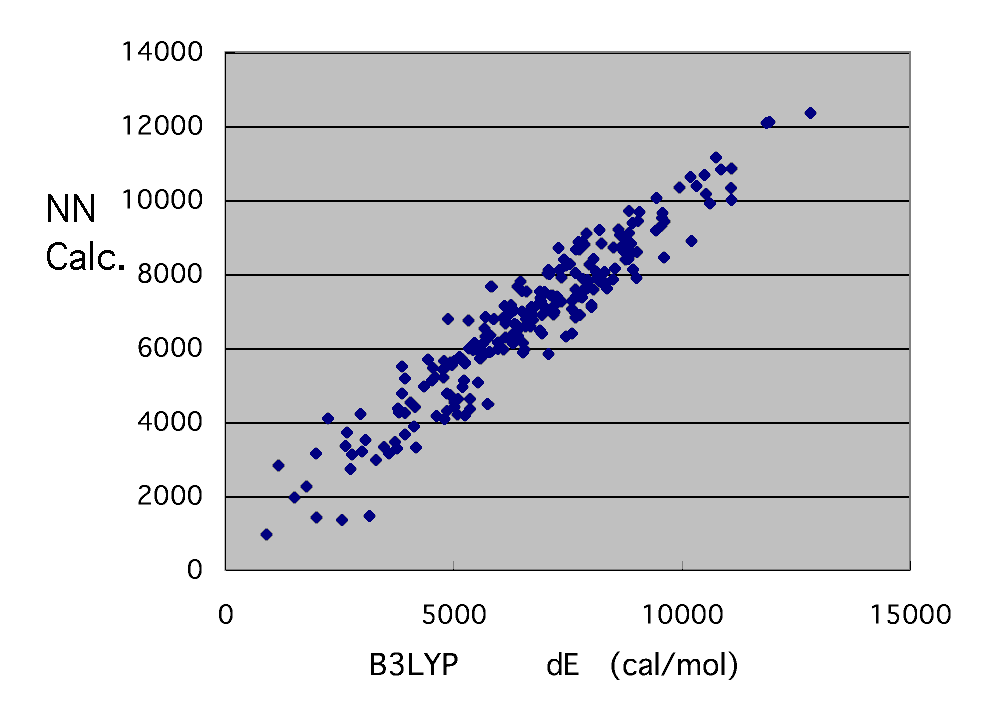
The result of the neural network who is going to reproduce calculation of this B3LYP / 6-31G** level,
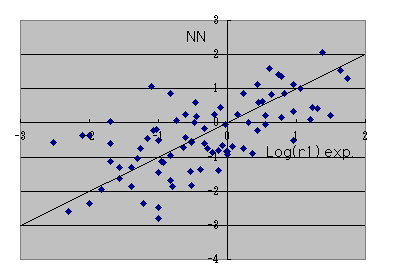
. I think that it is the accuracy from which it comes to swerve. Whatever it may call it, it is attractive that a reactant ratio can be guessed in a moment only by calculating by MOPAC.
It is calculation of B3LYP / 6-31G** level by this neural network. The applet guessed only from radical structure and monomer structure
In this way, that a rate of reaction can be guessed by a neural network means things as making it sublimate to a knowledge base, when a neural network analyzes the database of a certain semantic B3LYP / 6-31G** level. That is, if there are 1000 kinds of monomers, a database will collect 1 million transition states by 1000*1000. The knowledge of how MOPAC calculation of 1000 radicals and monomers was done, the SOMO, HOMO, LUMO, a charge, etc. combined, and the knowledge base has caused the phenomenon of activation energy or a frequency factor tends to be drawn, and it is going to guess the rest from the knowledge by about 3000 calculation.
reactivity of new monomer
Radical MOPAC calculation is only considered as a monomer to calculate a new monomer.
It comes out. Activation energy and a frequency factor are calculated in arbitrary combination. It can carry out.
Cyclohexene often used by photoresist as a new monomer
here A (cHn) trifuluoro ethyl-alpha chloro-acryrate (EBR), methacrylonitrile
(MAN) butadiene (BD) vinyl-2-pyridine (2PZ),isoprene (IsP), chloroprene(ClP),
tButyl acrylate(tBu), vinyl-4-pyridine (4PZ), methyl vinyl Ketone(keto),
alpha fluoroacrylate (FAM), cyano vinylidene (2CN), Isobutyrene (IB) and
trifluoromethacrylonitrile (F3MAN) were added. These monomer and a radical The neural network made to learn without incorporating
at all Work It was as follows when the system which is alike and
contains these monomers was made to guess.
The thing which dE does not suit :cHn monomer an EBR radical, BD radical, an IsP radical, a ClP monomer,2CN and IB
Frequency-factor does not suit: Add above and keto, FAM, and F3MAN.
dEA917 |
B3LYPdE |
NN |
Hindo |
NN |
cHn-cHn |
10140.6128 |
5868.305 |
3.6825E-16 |
2.2983E-14 |
chn-Mal |
2038.73577 |
2961.7627 |
1.4252E-15 |
4.7639E-15 |
cHn-MMA |
4978.88248 |
4417.3184 |
3.0198E-15 |
2.3229E-14 |
Mal-cHn |
9816.78878 |
7758.5107 |
4.182E-15 |
1.1433E-14 |
MMA-cHn |
15858.3667 |
9778.535 |
7.0742E-17 |
2.0964E-15 |
|
|
|
|
|
EBR-EBR |
8383.42803 |
5418.4893 |
8.4273E-16 |
1.9301E-13 |
EBR-MMA |
7815.4808 |
10444.751 |
1.3892E-16 |
3.6278E-14 |
|
|
|
|
|
|
|
|
|
|
Vac-MAN |
3716.21969 |
4117.8306 |
5.9933E-15 |
9.1537E-15 |
MAN-MAN |
9978.70081 |
9078.901 |
1.5093E-15 |
2.7545E-15 |
MAN-MMA |
9661.15241 |
9673.638 |
1.7624E-15 |
1.2623E-15 |
MAN-St |
9029.19345 |
9092.24 |
3.9633E-16 |
5.3786E-16 |
St-MAN |
8724.19637 |
8584.823 |
4.074E-15 |
4.5989E-15 |
MAN-Vac |
11492.39 |
10677.743 |
1.2707E-15 |
1.6353E-15 |
MMA-MAN |
8419.82686 |
8788.75 |
6.8941E-16 |
5.8944E-16 |
2PZ-MAN |
8721.68611 |
8870.69 |
7.3631E-15 |
2.9011E-15 |
|
|
|
|
|
MAN-BD |
8427.35765 |
9269.17 |
1.2242E-15 |
7.4706E-16 |
BD-MAN |
10917.5395 |
5923.492 |
1.2155E-14 |
3.2982E-14 |
|
|
|
|
|
2PZ-2PZ |
8058.97641 |
6544.577 |
7.9006E-15 |
1.2262E-15 |
2PZ-AM |
8295.56879 |
8610.197 |
6.0478E-15 |
2.6665E-15 |
2PZ-AN |
7738.29019 |
8716.495 |
6.5814E-15 |
4.0184E-15 |
2PZ-Vac |
11702.6246 |
12431.175 |
1.2229E-14 |
2.9756E-15 |
Vac-2PZ |
6154.3136 |
2964.3513 |
9.0277E-15 |
3.0643E-15 |
AN-2PZ |
6391.53355 |
4609.5386 |
5.4058E-15 |
3.0607E-15 |
|
|
|
|
|
IsP-AN |
11183 |
6342.667 |
1.4527E-15 |
2.3447E-15 |
AN-IsP |
7137.08196 |
8026.433 |
1.8346E-14 |
1.9626E-14 |
IsP-ClP |
11563.9325 |
8257.195 |
9.6587E-16 |
1.4262E-15 |
IsP-BD |
12533.522 |
7929.1313 |
1.1042E-15 |
9.4508E-16 |
|
|
|
|
|
Vac-ClP |
2838.25486 |
5574.0967 |
1.3287E-14 |
2.9369E-15 |
MMA-ClP |
7423.25205 |
8939.393 |
4.6628E-16 |
3.3919E-16 |
AM-ClP |
4546.48951 |
6601.5835 |
8.6019E-15 |
2.7243E-15 |
AN-ClP |
6587.96171 |
7542.2295 |
1.0394E-14 |
2.2687E-15 |
|
|
|
|
|
tBu-tBu |
6678.33121 |
7466.934 |
9.2113E-15 |
4.3796E-15 |
4PZ-4PZ |
8665.83273 |
8709.371 |
1.0167E-15 |
2.3383E-15 |
KeTo-KeTo |
7298.99399 |
6928.032 |
2.9986E-15 |
7.7817E-16 |
FAM-FAM |
6951.32242 |
6500.1807 |
2.6427E-15 |
6.5849E-16 |
|
|
|
|
|
2CN-2CN |
11789.2287 |
2175.6362 |
1.3607E-15 |
4.3563E-17 |
IB-IB |
9450.29023 |
6482.1396 |
1.8816E-15 |
1.4896E-15 |
F3MAN-F3MA |
7886.39576 |
6518.2036 |
1.4595E-16 |
3.1912E-13 |
As for one with a clear cause, F3MAN and 2CN are radicals among these things not suiting.
It is extrapolated by LUMO.
tBu, EBR, and cHn are extrapolated by XXX.
Moreover, butadiene, isoprene, isobutylene (chloroprene), cyclohexene
is not contained in the first study data.
Since the result obtained this time is fed back immediately Combination of 32*32=1024 It is alike, it attaches and can calculate now freely.
| Top page Frequency View of TS Edit TS Structure How to Use Check Frequency How to use |
MOPAC Calculation Ab initio calculation Monte Carlo simulation Polymer properties estimation |
