Abstract of HSPiP | How to use Y-Predict
2012.Dec.19
Y-Predict is Thermo-Chemical properties estimation program. These estimation schemes were developed by pirika.com. The indivisual theories and correlations are listed on pirika.com.
The input format for chemical calculations in HSPiP are SMILES, InChI and MOLfile. There is no GUI input of molecular structure.
Now, we present you HTML5 base software, "Y-Predit" as a Power Tool of HSPiP.
Browser support:
| Mac | Windows | Linux | |
| Chrome (ver. 23) | ◯ | ◯ | |
| FireFox (ver. 17) | ▲(Web Storge X) | ◯ | |
| Safari(ver. 6) | ◯(OSX, Lion+) | ? |
|
| Opera (ver. 12) | X (File API X) | ◯ | |
| IE (ver. 10) | - | ▲(Web Storge X) |
IE 6-8 does not support HTML5.
IE-9 does not support FileAPI so can not validate license file.
IE-10 has a problem in Local Storge and users need to validate every time.
Opera for Mac does not support FileAPI.
FireFox for Mac has a problem in Local Storge and users need to validate.
We strongly recommend Chrome browser.
Run the program
Please open index.html with an adequate HTML5 browser. (By clicking with right button and select open with application or drop index.html to alias of browser icon or, from HSPiP, clicking the PowerTools menu option) Then you will see the next startup screen. The language (English or Japanese), Button name and appearance are dependent on your browser. If you change size of browser window, the Canvas size is also change according to Window size.
How to Draw molecules is the same as Draw2Smiles, so pictures are from that help file.
Start up screen.
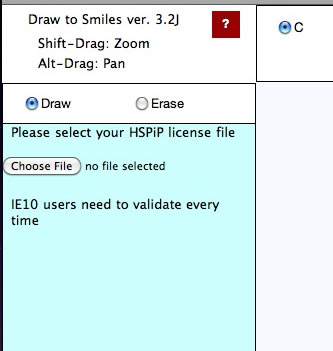
Validation
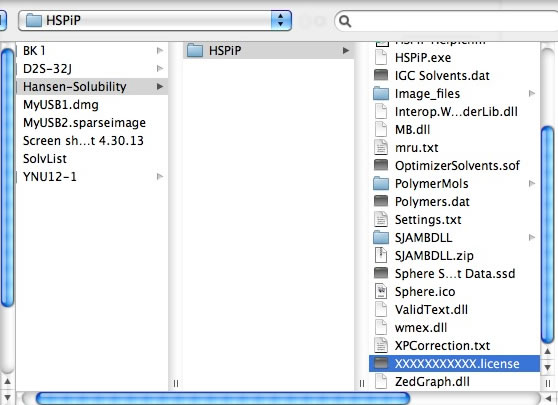
The first time you run a Power Tools, you need to register your HSPiP License File to the browser. Please click Choose File button (Button name and appearance are dependent on browser) and select your HSPiP License File, typically in c:\program files (x86)\Hansen-Solubility\HSPiP.
Once you open the License File, the browser stores the information locally and every Power Tools will run without verification. (IE10 does not handle local storage properly and users need to validate every time)
Drawing
The light blue part is called the canvas in HTML5. The actual drawing is done on this canvas. Other buttons, radio buttons, text fields, text areas previously existed as a form object. CSS controsl position, size, color and appearance in HTML5. Therefore, if your browser does not support HTML5, you cannot see the canvas, and position of the parts looks a mess. If you change browser window size, the Canvas size also changes automatically.
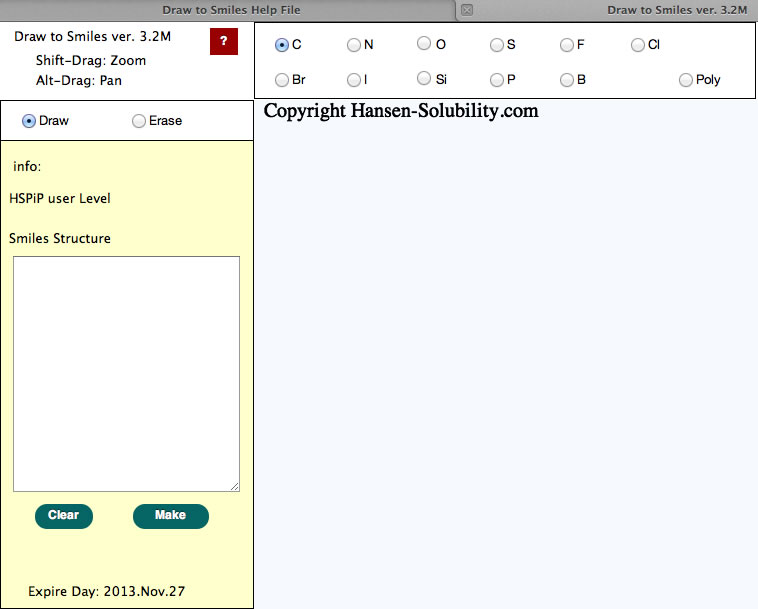
Above picture, using a carbon atom, Draw mode is selected.
Then, press the mouse button inside the canvas and drag (without releasing the mouse button or finger) the position, you will see the drawing like below.

While pressing the mouse left button, you will see the atom that you selected with the radio button, and will see the bond. If you move the mouse, the bond will re-draw at 15 degree intervals.
If the position where you release the mouse button is not identical to the first atom, the program put atoms on either end of bond. When the mouse moves near to an atom, that atom is detected an a pattern appears to show this. If you click when the pattern appears, you will select that atom. If you click before the pattern appears, a new atom will be created. So if you want to draw a large molecule, do it very slowly. It needs time to check if the mouse is pointing at an atom.
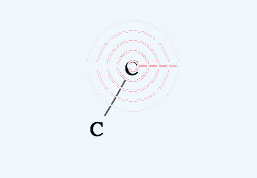
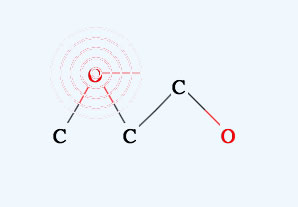
If you select another atom with a radio button, that atom will be used in drawing or for replacing.
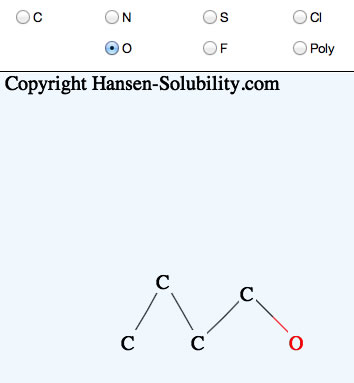
If you click the mouse on one atom, and release immediately, that atom will replace the selected atom.
Please select Erase mode. The background color is changed to remind you that you are in a different mode.
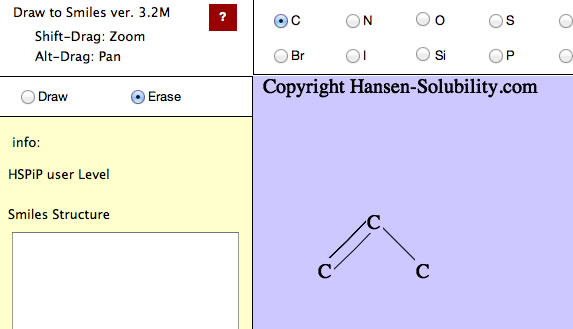
If you move the mouse on to an atom, (the pattern will appear), then left click and release, the atom will disappear. If you move the mouse between two atoms, two patterns will appear, then left click and the bond will disappear.
 →
→
Set Draw mode again.
If you want to make a double bond, move the mouse on the first atom (confirm that the pattern appears) then left click and drag to the other atom. When you see the pattern, release the mouse button and you will get a double bond.
 →
→
When you Alt drag (option key for Mac) you can move the molecule inside the canvas. If you Alt+Drag on one atom, you can move only that atom.
 →
→
Shift+Drag Zooms the molecule.
 →
→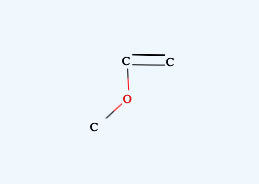
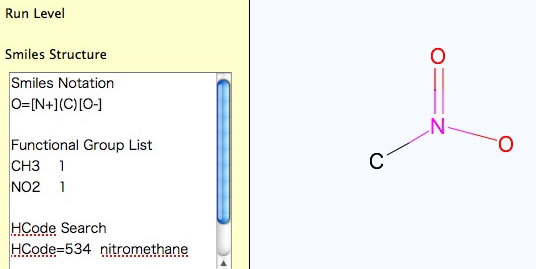
For Nitro Compounds, please write like below.
It is very important to be careful with the mouse moves, if you want to build very large molecule. Be patient and confirm that the pattern appears before clicking.
(On a touchpad remember to unclick before you release the Alt or Shift key, otherwise you will draw a new atom by accident)
When your structure is complete, click the Predict button, you will get the properties of this molecule.
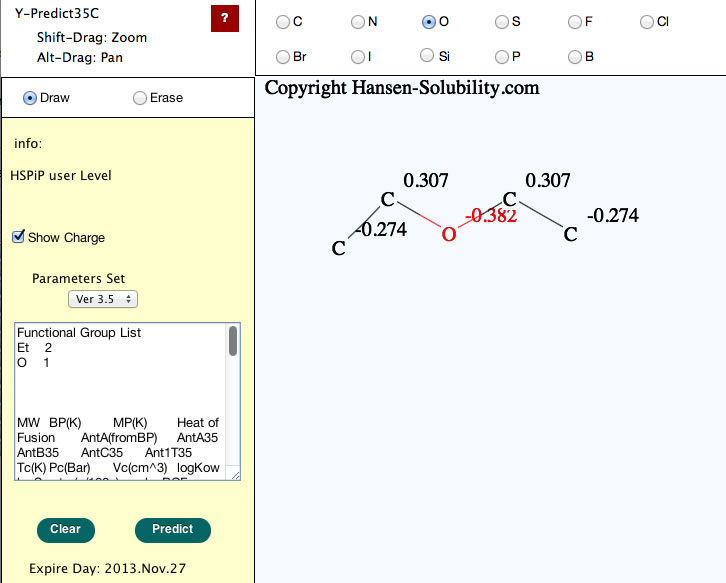
Please check the functional group list so as to confirm that the molecular break by computer is correct.
Please select the calculated results and copy(Ctr-C).

You can paste(Ctr-V) to spread sheet and arrange as you like.
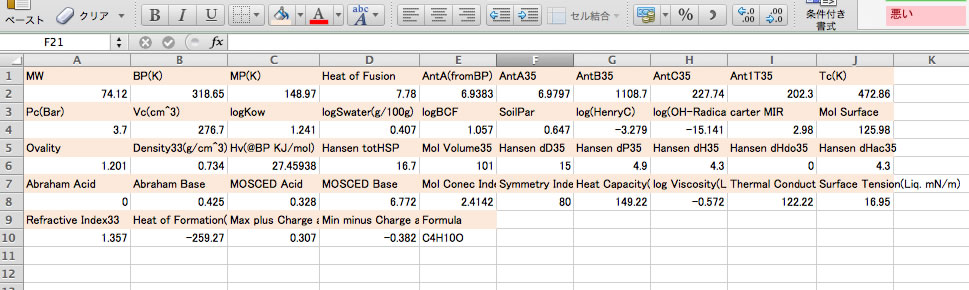
Ant1T35: The temperature that vapor pressure become 1mmHg
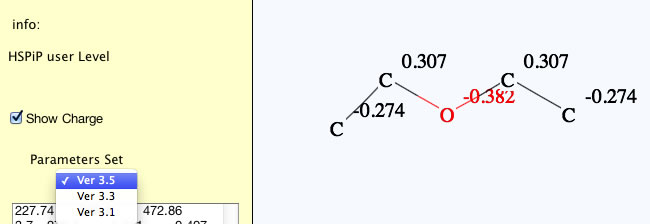
You can choose a parameter set with pulldown menu, so if you want to calculate with previous version parameter sets (for historical consistency), please select that version. HSPiP 3rd Edition used Y-MB Ver 3.1. Y-Predict calculates atomic charge with 2D-QEQ method. (Program automatically adds hydrogens for QEQ calculation, but hydrogens are not displayed in this version. If I get feed back from many users that they want to see the hydrogens, I will increase the priority of programing.)
For Nitro Compounds, please write like below.
The maximum atom number is set to 120 as heavy atom (same with HSPiP), but these thermo-chemical properties estimation become meaningless if the boiling point become above 700K. Please use carefully to that chemical's result.
For the next version of Y-Predict, I will build several estimation schemes according to chemical types.
New version (2013 Aug. 15) of Y-predict calculate temperature dependent properties.
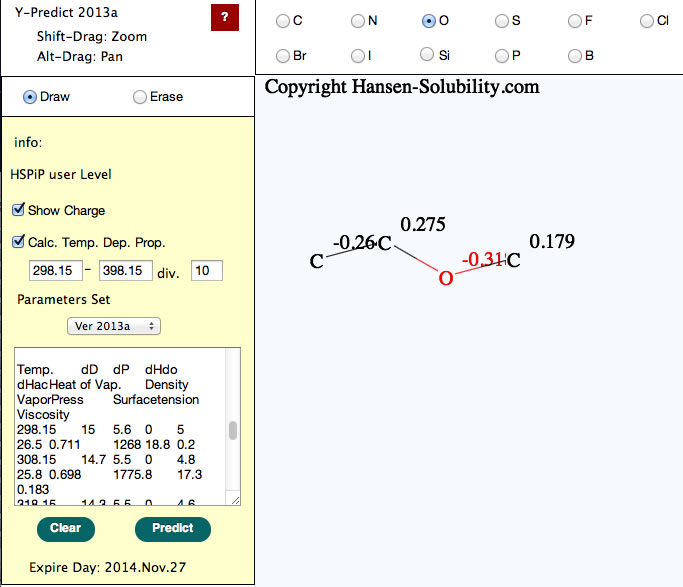
Please set start temperature and end temperature with division. program will calculate temperature dependent of HSP, Heat of vaporization, density, Vapor pressure, Surface tension and viscosity.
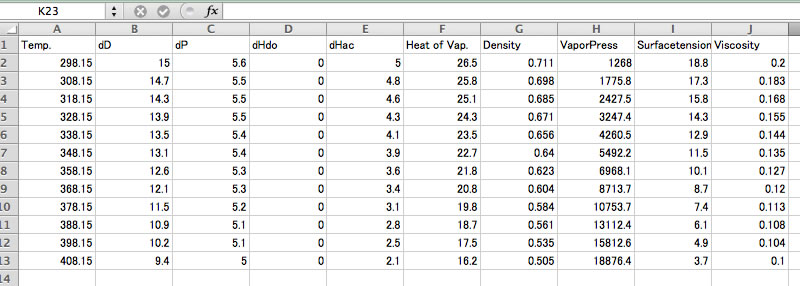
Please copy& paste to spreadsheet for further analysis.
Caution: The calculation routine of temperature dependency of HSP is completely defferent from HSPiP method. This method calculate Hv(T) and Vol(T) and get tot HSP(T)=sqrt((Hv(T)-RT)/V(T)).