
2020.12.27
PirikaでSTEAM > デジタル分子模型で見る化学結合> 1. 分子構造の調整。
試しにこれらのページで電子書籍を作ってみました。
epub3のビュアーを持っているなら試してみるのも良いでしょう。
1. 分子構造の調整。
A. 二面角(ねじれ角)の変更
例えば、ジメチル・パーオキサイド(CH3OOCH3)の3次元の座標データがあったとしましょう。
その座標を元に分子を表示させると次のようになります。
キャンバス(表示エリア)内でマウスボタンを押しながら動かす(ドラッグする)と分子が回転します。(iPadなどではキャンバスをタッチしたまま動かします。)
Alt key(Option key)を押しながらドラッグすると画面上の位置を変える事ができます。
(iPadなどでは、指を3本タッチさせて動かします。)
真ん中の二つの酸素が重なるように分子を回転してみましょう。
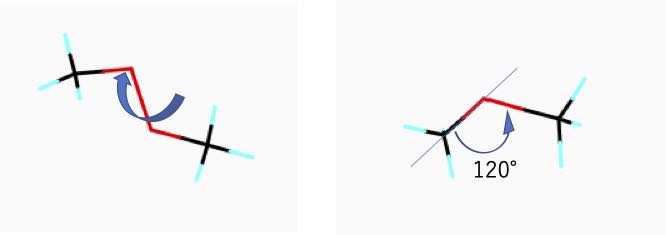
酸素と酸素の間の結合は回転させる事ができます。そこで、その結合がどのくらい捻れているかを2面角(Tortional Angle)で表す事が行われます。
この2面角というのは、原子を4つ定義した場合に、最初の3つが作る面と後の3つが作る面のなす角度で表されます。
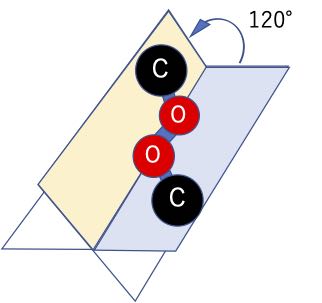
この表示プログラ中では、炭素-酸素-酸素-炭素に順番に1-2-3-4と割り振っているので、プログラムは自動的に2面角の計算を行い、120°である事が示されています。
(2面角は通常の角度0°-360°とは異なり、-180°から180°で表します。)
ここで、Shift keyを押しながら、マウスを左右に動かしてみてください。
(iPadなどの場合には、二本指でタッチして指の間隔を変えます。)
2面角を色々な値に変える事ができます。
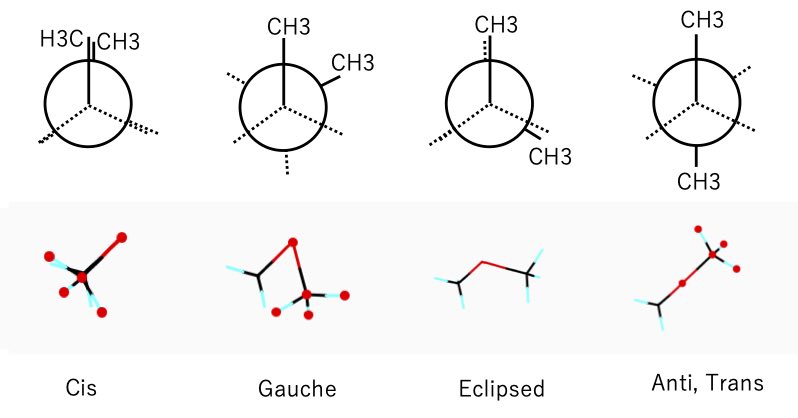
このような図はニューマンの投影式と呼ばれています。
60°おきの構造に、シス(C)、ゴーシュ(G) 、エクリプス(E)、アンチ(A)という名称がついています。
(ただし、エクリプス=太陽が月で隠される日食のことで、シス体もエクリプスです。)
このように、単結合の回転や、孤立電子対を持つ原子の立体反転によって生じる異性体を、立体配座異性体といいます。
n-ブタン(CH3CH2CH2CH3)の真ん中の単結合を回転させた場合には、エネルギーは次のようになります。
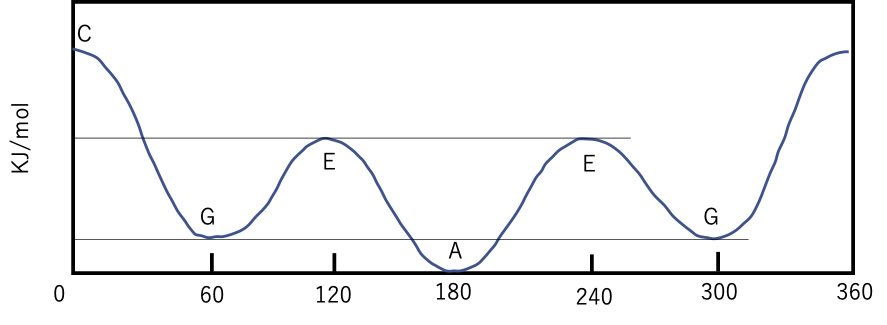
通常はシス構造を持つ化合物がエネルギーが最も高くなり、不安定になります。そして重なり具合の大小によってエネルギーが変わります。こうした山の高さを回転障壁と呼びます。
低温では一番安定な配座に落ち着き、熱をかけていくと回転障壁を乗り越えて、エネルギーが高い配座の割合が増えます。
分子が複雑になってくると、このような配座数は非常に多くなります。最も安定な配座を求める配座解析は、医薬品の解析などで行われています。
しかし、2,2,3-トリメチルブタンなどでは、非常に高いであろう回転障壁(メチル基2つがエクリプス)が1種類と安定な構造1種類しか無いことは、次のモデルで分子の部分回転(シフト・ドラッグ)をしてみるとわかるでしょう。
分子力学計算では、結合長、結合角、2面角の振動をバネで表します。様々な原子種の組み合わせについて、異なったバネ定数を与えることによって、この回転障壁を見積もる事ができます。
分子軌道計算で、このような回転障壁を計算するには、分子構造の最適化(一番安定な構造を自動的に探してしまう)をオフにして、計算を繰り返します。
B. 結合長の変更
分子を組み立てた時、結合長を変更したい場合があります。
分子の安定構造を求めたいのでは無く、不安定構造、例えば遷移状態などを求めたい時です。
遷移状態というのは、
A + B → C
という反応があったときに、通常はエネルギーの高い山(反応障壁)があって、その山の頂点での構造を言います。
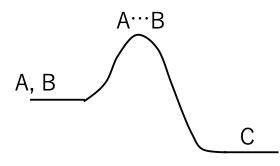
A,Bは個別の分子ですので、AとBの距離は任意です。
Cは結合して一つの分子になっているので、AとBの間は通常の結合長になっています。
遷移状態では、通常の結合よりはやや長い(約1.4倍程度)結合になります。
もちろん、結合角もsp2 からsp3 へ変わったりしますが、分子軌道法を用いて遷移状態を求める時の初期値としては、Cの構造を作り、結合長を変更するという事がよく行われます。
Shift+ドラッグすると、メトキシラジカル(CH3O・)がプロピレンに反応する際の結合距離を変更する事ができます。そして、できた構造を取り出して分子軌道計算の初期構造にします。
C. 結合角の変更
結合角を変更したい場合はあまり多くないと思います。
例えば、カルボン酸のナトリウム塩の構造を作るときに、通常の酸の水素をNaに変更した左の構造にするか、酸素は等価で結合次数が1.5である右の構造にするかは、意見の別れるところでしょう。
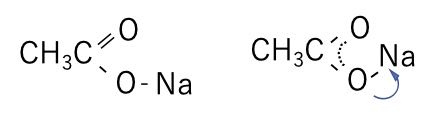
Shift+ドラッグして、OCO面内でNa原子を動かす事ができます。
現物の分子模型にも、長さを測るスケーラーが付属していますが、様々な測定はデジタルの方が優れていると言えます。
ここでの例題では、どこを動かすかの原子をあらかじめ設定してあります。
マウスで、選択された原子をクリックするとクリアーされます。
長さの場合は原子を2つ。
角度の場合は原子を3つ。
ねじれ角の場合は原子を4つ選択すると、他の部分を動かす事ができます。
1.分子構造の調整。
2.分子集合体の分子模型作成。 ちょっと計算が重いので注意
3.各原子上の電荷を計算。 ちょっと計算が重いので注意
4.ある温度における分子(原子)の運動。
5.π結合とσ結合の違い。
6.作られる直前の化学結合の様子。
7.HOMO-LUMO遷移エネルギーと化学結合。
8.振動解析結果のアニメーション。
9.デジタル教科書の作成。
試しにこれらのページで電子書籍を作ってみました。
epub3のビュアーを持っているなら試してみるのも良いでしょう。
10.豊かな化学のために。
12.全フッ素化キュバンのLUMOが電子を閉じ込めた!
Webアプリ版、CNDO/2を使った教材作成について
「こういう教材を作りたいので、pirikaのアプリを使いたい」と言うご要望がありましたら、メールを頂ければ幸いです。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。