
Pirikaでやっている事は難しく、ハードルが高いと言われることが多い。
でも、高いハードルを無理に飛び越えるのは老体には無理なので、くぐるのも大事だ。
例えば、ダイオキシン類(ポリ塩化ビフェニル)の毒性のQSARなどは難しい。
女性ホルモンのEstroneと最も毒性の高いダイオキシンのPCDDをY-MBで計算すると次のようになる。
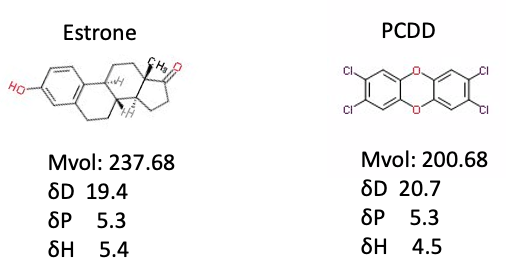
分子の形も、ハンセンの溶解度パラメータもよく似ているので、女性ホルモンを受け入れるレセプターに入り込んで悪さするのかと思う。
ここまでは、HSPiPソフトを購入すれば誰でも計算できる。
しかし、Y-MBの開発者としては、方法論の限界にも直面する。
例えば、原子団を数えようとすると、分子をバラバラにする必要がある。バラバラにすると原子団がどこに付いていたかの情報は消えてしまう。特に芳香属化合物で困る。
PCDDの場合だと、左のベンゼン環も右も、4置換のベンゼンとしてしか認識されないので、塩素の位置が変わっても計算値が同じになってしまう。
塩素の位置が変わるとEstroneとは見かけは変わるし、毒性も変わるのでY-MBをどうにかしなくてはならない。
非常に高いハードルになる。
現在、ver.6用のY-MB(2021)の構築と評価を進めている。
これを、ダイオキシン化合物に適用してみよう。
Y-MBは化合物のSMILES構造式があれば物性値を推算する。
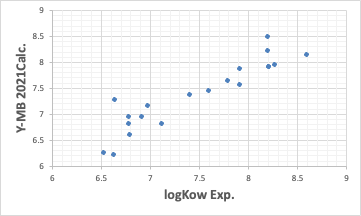
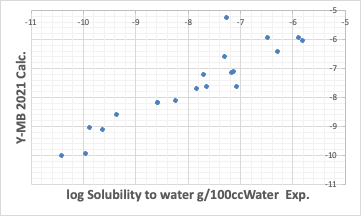
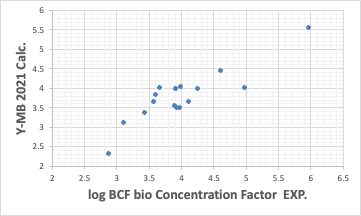
ここまではそこそこの性能を示している。こうした熱力学的物性値推算が強いのがY-MBの特徴だ。
ところが、ダイオキシンの毒性当量因子TEF (toxic equivalency factors) をY-MBの識別子を使ってQSAR解析すると、どうしても計算が合わないものが出てくる。
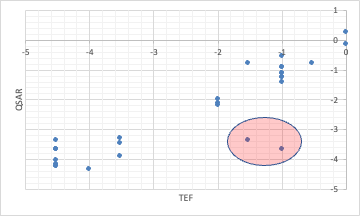
これは、メタ位、パラ位に塩素を持つPCBはコプラナーPCBと呼ばれ毒性が非常に高くなるからだ。
そこで、ハードルをくぐる方法を探す。
HSPiPにはVer. 5.1あたりからRDKitが搭載されている。これはトポロジカルな識別子や3次元構造を作成する。そこで、Y-MBに加えトポロジカルインデックスと3次元構造からCNDO/2分子軌道計算した結果も含めてTEFを推算するQSAR式を構築する。
つまり、全部自前でやることを諦めて、ハードルをくぐると、視界が広がる。
溶解性しなければ、毒も毒ではない:HSPの溶解パラメータ
分子の形:トポロジカル・インデックス Chi3n
電子的な情報:HOMO
分子の安定性:Heat of Formation
から新しいモデル式を作成する事ができる。
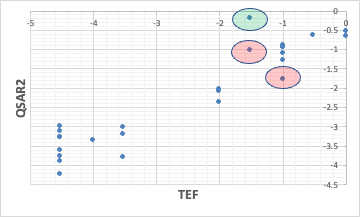
先ほど外れていたコプラナーPCBは良くなったが、逆に大きく外れるものが1つ増えた。
それが何故外れるのか考察する事が大事なのであって、自前でソフトを開発する時間をそちらに振り分ける事ができる。
通常のAI/MI/MLはどれか一つだけで行うことが多い。
でも、one stopで必要な識別子を全て調達できると、きっと地平線が広がる。
次の図は大きく外れた1,2,3,7,8-PeCDFをCNDO/2で計算したものだ。(計算結果を表示しているのではなく、ブラウザーの上で計算しているので、速度の遅いマシンでは計算に時間がかかる。)
分子を回転:マウスでドラッグ(マウスボタンを押したまま動かす) iPadでは指一つで押さえて動かす
位置を動かす:Alt(MacではOption)キーを押しながらドラッグ。 iPadでは指3本で動かす
拡大・縮小:Shiftキーを押しながらドラッグ。iPadでは指二本で横に広げる、狭める。
今の時代は、プログラミングと言っても、全てを自分で書く必要はない。でもpythonでライブラリーを結びつけるだけをプログラミングと間違って認識してもらっても困る。
アルゴリズムを考えられるようになるのが大事で、意外とそれは楽しいことだ。