
2018.12.13
情報化学+教育 > 情報化学 > RDKitの使い方
RDKitの使い方
Chemo-Informatics用のフリーウエアーの代表といえばrdkitでしょう。
RDKitを本格的に使う場合にはPythonなどの知識が必要ですが、ブラウザーだけでできることも多いです。
RDKitのJavaScript版、RDKitjsを使って実際にマテリアルズ・インフォマティクスをやってみましょう。
分子の構造データ準備
計算を行うためには、分子のSMILESの構造式が必要になります。SMILESの構造式がわからない場合にはこちらを参照してください。
JSME(Webアプリの分子のお絵かきソフト)を使って](https://www.pirika.com/Education/JP/ChemoInfo/FreeTools.html)分子の構造をお絵描きして、Smilesの構造式のリストを作っておくのもよし、ネットでCAS番号や分子の名称などからSMILESを検索しても良いので、とにかくまずはリストを作りましょう。
化合物の名称などがある場合に、その化合物のCAS#、Pubchem番号を調べたいならこのサイトがおすすめです。
一括で変換する機能(Batch)もあるので、化合物の名称の列をペーストするとCAS番号などに変換できます。
Pubchem CID, CAS#などがあるのであれば、このサイトでSMILESに変換できます。
今回使用するデータ
上の化合物群は、ある論文(Jpn J Ind Health, 1994;36:314-323)から取ってきた化合物のラットの経口急性毒性のデータです。
logKow(オクタノール/水分配比率)、logS(水への溶解度, g/100ml水)、logBCF(生物濃縮性)のデータは私が付け加えました。
このデータを表計算ソフトにペーストして化合物のSmilesの構造式を用意しましょう。
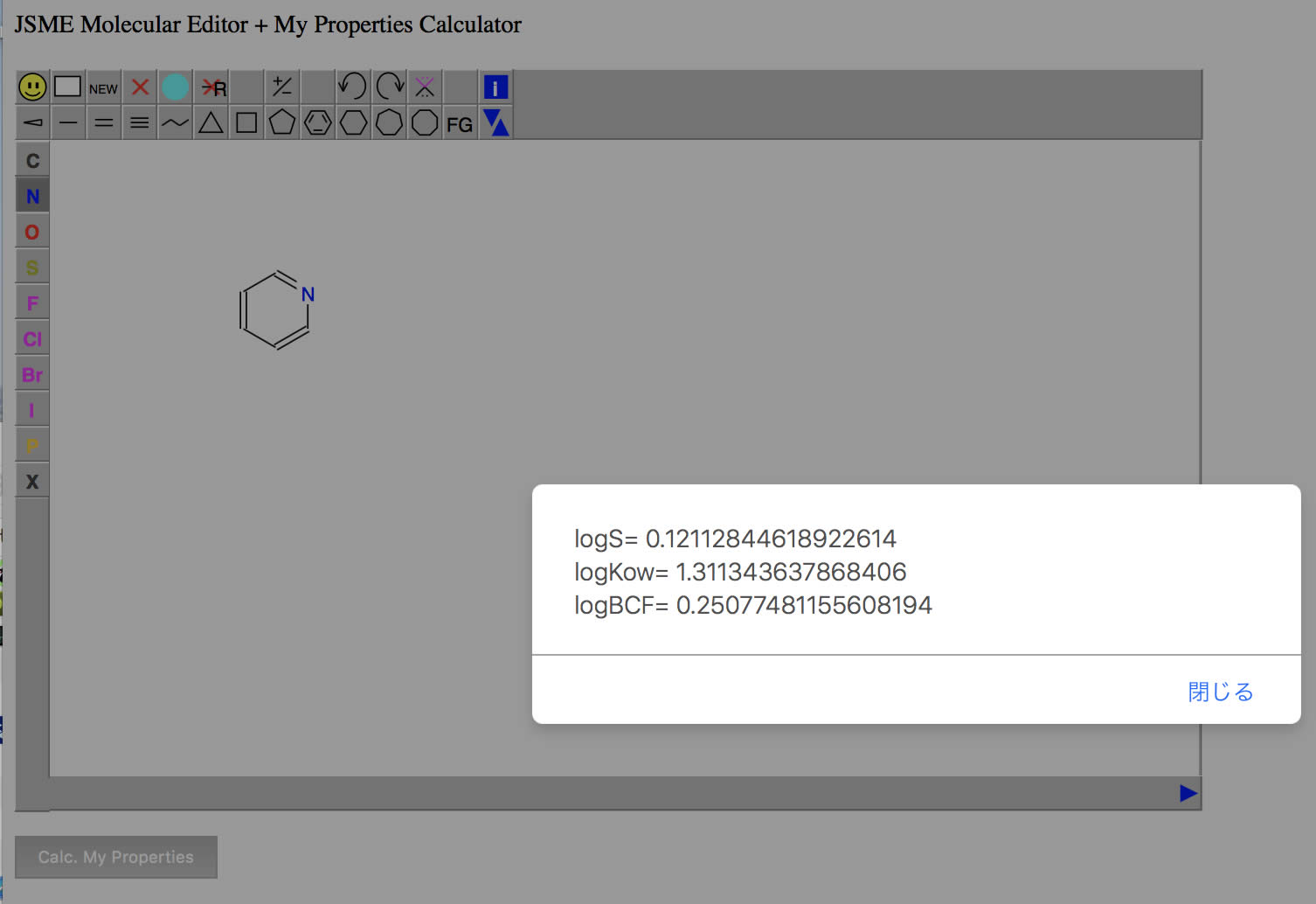
JSME(分子のお絵描きソフト)
JSMEを使って分子構造を描き、Smilesのリストを先に作ってしまうならこのバージョンを使います。どのみち後で使うので、使い方は慣れておいた方が良いでしょう。
RDKit(Webアプリ版)を使って計算する。
JSME-RDKit連携バージョン
JSMEで2次元構造を描画しながら、RDkitを使って3次元座標とDescriptorsを生成するならこのバージョンを使います。
RDKit ブラウザーバージョン
Smilesのリストがすでにできているのなら、順番に1つずつRDKitで計算します。
どのバージョンを使っても良いのですが、RDKitjsは6Mbyteを超えるファイルサイズなのでネットワークのスピードとマシンのスペックはそれなりのものがあるという前提はご理解しておいてください。
Smilesのリストができているとして、ブラウザーバージョンのRDKitを使って説明を続けます。
リンクにアクセスすると次のようなページが現れます。

2022.4.19
WindowsでこのWebアプリを使うと、エクセルからペーストした時に改行も加わってしまうことがわかりました。結果が乱れるので修正しました。
2019.1.9:分子結合インデックスの計算に誤りがありました。
本来、Chiパラメータは水素原子は計算に含めないはずです。
Smilesの構造から3次元分子を作る際に水素を付加させていました。
この付加の影響でRDKitの計算結果のうち特にChiパラメータが大きくずれてしまっていました。
プログラムの方は修正しましたが、このページにある計算結果、選択された変数は少し変わります。
ここでは、まず、RDKitが吐き出す識別子として何を選択するかをチェックボックスで指定します。
大事なのは途中で変えないことです。
テーブルのカラムが変わってしまいます。
(ToDoリスト:計算が始まったらチェックを変えられないようにする)
そして一番最初の化合物のSMILES式をペースとしてAid to Fileボタンを押します。
最初の計算はRDKitの読み込みに時間がかかりますが、2回目からは早くなります。
そして次々にペーストしてはボタンを押すを繰り返します。
(プロ版ではSmilesを一括してペーストでき、連続計算できます。識別子は117種類吐き出す。)
RDKit計算結果の取り出し
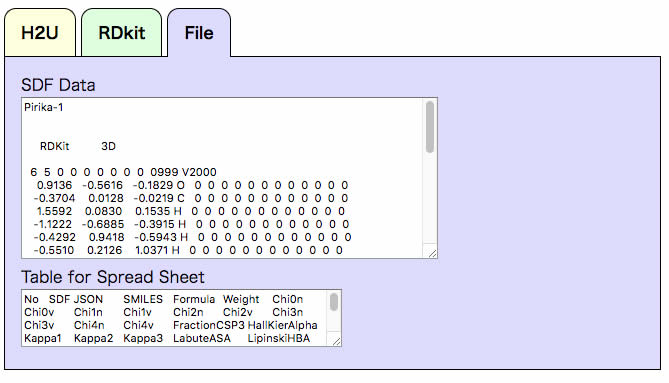
計算が終了したら、Fileタブを開いてみましょう。
このプログラムは2種類のファイルを生成して、テキストエリアに貼り付けてあります。
SDF Dataは分子の3次元座標と幾つかの値をSDFフォーマットで書き出します。
分子は$$$$で区切られています。
この3次元座標は分子軌道計算に使ったり、RDKitで複数の分子を計算するのに必要となるフォーマットになります。
テキストエリアの中で”全てを選択”してテキストエディターなどにペーストしてセーブしておきます。
テキストエリアは右下の部分をつまんで動かすと大きさを変えることができます。
(HTML5のすごい機能です)
もう一つはタブ区切りのテーブルのファイルです。
1行目は識別子の種類で2行目以降は各分子の識別子の値が入っています。
これも全てを選択して表計算ソフトにペーストします。
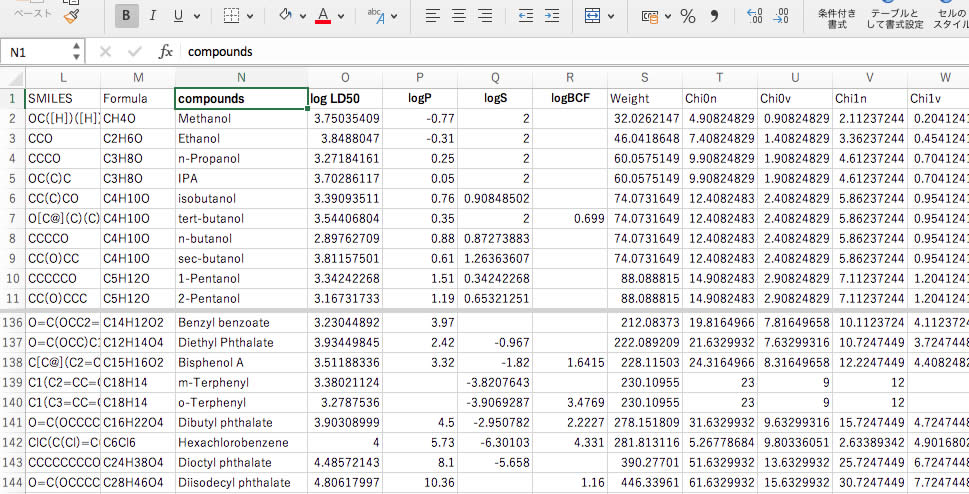
Smilesのリストがあれば、15分もしないで、上のような物性値とRDKitの識別子のテーブルができあがります。
RDKitjsはオリジナルのRDKit(Python C/C++)のサブセット版なので、オリジナルのものと比べると算出される識別子の種類は少ないです。
(ToDoリスト:新しいJavaScriptバージョンが出たら更新する。Finger Printデータは出力に含めていない。必要であれば組み込む)
物性推算式の作成
ここまで出来たら、次は物性推算式の構築に取り掛かれます。
まず、化合物の水への溶解度の推算式を構築してみましょう。
この例題はRDKitのデモとしても有名で、RDKitの使い方の例題としてもネットに様々情報が溢れています。
例えば
Kerasで化合物の溶解度予測香川大学
1000個以上の識別子を使って、ニューラルネットワーク法やPLS法を使ってモデルを組み立てています。
しかし、こうした方法を使いこなすにはプログラム言語Pythonの知識が必要になります。
もっと簡単にWebアプリでやってみましょう。
我々、化学者が推算式を作るとなると、一番わかりの良いのは重回帰式でしょう。
重回帰式では
物性値=a+b*識別子1+c*識別子2+ +n*識別子x
という式のa, b, c, nの係数を求めた式です。
これは中学だかで習う連立方程式](https://www.pirika.com/Education/JP/MAGICIAN/Beginner/index.html)の解と同じ事になります。
この重回帰計算の機能は表計算ソフトにも搭載されているので使った事があるかもしれません。
ここで問題になるのが、今回RDKitから吐き出された識別子が37種類(1000以上と比べれば少ないですが)もあるということです。
それを全て使って式を立てるのは共線性(説明変数同士に相関がある)の問題などがあり、得策ではありません。
変数が多い場合、教科書的には、主成分回帰やPLS回帰を使うように勧めているのですが、この後説明するように、それはそれで問題があります。
ここでは、オーソドックスに授業でいつも使っている変数選択重回帰法を使ってみます。
この変数選択重回帰法はYNU-YSBプログラムに搭載されています。
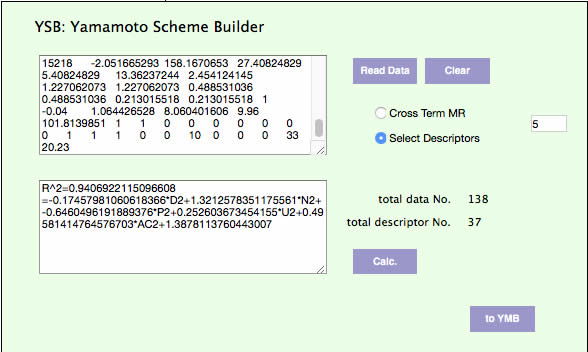
データを上のテキストエリアに貼り付け、「変数を5つ選択してR^2 の高い重回帰式を作成しない」とやると例えば下のテキストエリアにあるような答えが出ます。
この=の後ろにある式は表計算ソフトに貼り付ければすぐに結果を見ることができます。
(ペースト先は2行目のみなのは注意!)
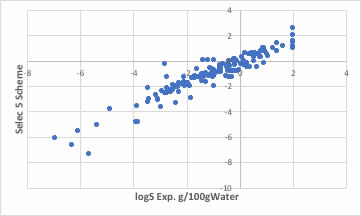
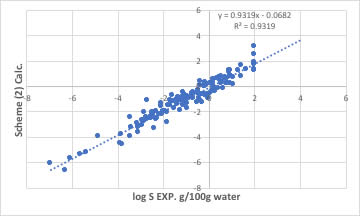
簡単に水への溶解度を表す重回帰式が得られました。
logS=-0.17457981060618366*Chi0n+1.3212578351175561*FractionCSP3+-0.6460496191889376*Kappa1+0.252603673454155*LipinskiHBD+0.49581414764576703*NumHBA+1.3878113760443007 (1)こういう事が簡単に出来てしまうので、Materials Informaticsという技術は怖いのです。
この式を組み込めば、JSMEで分子のお絵描きをした後、裏ではRDKitを走らせ、この5つの識別子を計算して、水への溶解度を返してしまう事ができます。
特に大事なのが、自分が興味のあるデータセットから推算式を構築すれば、自分の興味のある化合物に特化した推算式になるという事です。
自分が高分子合成用のモノマーにしか興味がなければ、モノマーばかりから作ったテーブルで同じことをすれば、モノマーに強い推算式になるのです。
医薬品でも同じことです。これが今世界中の研究者がRDKitを使ってMIをやろうとしている理由です。
データを1000個,10000個とビックデータを集めてどんな化合物でも計算できるようにディープラーニングさせたいというのは、学者に任せておいて、企業では取り敢えず自分に関連する化合物群で式を作ってしまえばいいのです。
(ちなみにRDKitに含まれる1000データを解析するとこうなります)
YMB24Pro4MIで変数選択重回帰法をつかう
v-tubeで使い方のビデオを作った。
化学知識を使う
ここまでなら、化学の知識の無いデータサイエンティストにもできます。
私が主成分分析やPLS回帰はもう少し後で良いと言う理由を少し考えてみましょう。
先ほどの推算式は決定係数0.88(相関係数Rの2乗)なのでそれなりに良いモデルなのですが、それでは、このモデル式に合わない化合物はどんな化合物でしょうか?
誤差の大きなものをソートして見ると、Nitroanilineのo-,m-,p-は皆大きな誤差になっています。
何故かと言うと、式(1)で使われている、Chi0n、FractionCSP3、Kappa1、LipinskiHBD、NumHBA の5変数は、o-,m-,p-Nitroanilineで皆同じ値になってしまうからです。
ベンゼン環についた官能基の位置が違うだけなので、分子量を始めRDKitの吐き出す識別子の値がほとんど皆同じになってしまいます。
そこで、ベンゼン環についた官能基の位置によって値の変わる識別子を探します。
すると、Chi4nなどは値が変わってくるので、それを組み込むことにします。
logS=-0.2183895636547375*Chi0n+1.1113943882657598*FractionCSP3+-0.6154433219798137*Kappa1+1.0678749094594537*LipinskiHBD+0.5755342165654573*NumHBA+-1.5559623499363546*Chi4n+-0.45241634258826235*LipinskiHBD*NumHBA+0.08721946861665164*Chi0n*Chi4n+1.9585849028724098 (2)実際にやってみたところ、Chi4nを入れるだけでは改善せずに、YSBのもう一つの機能、クロスターム重回帰法を使うと式(2)が得られます。
LipinskiHBDとNumHBAと言うのは、HB(Hydrogen Bonding:水素結合)のD(ドナー)とA(アクセプター)の値です。
LipinskiのHBAとNumのHBDの値も識別子の中にはあるのですが、相関係数の大小だけで識別子を選んでいるので、たまたま、HDA, HBDとしてこれを選んでいただけです。
(ToDoリスト:変数選択で意味のあるペアはペアで導入する)
式(2)の中で、クロスタームでLipinskiHBD*NumHBAが導入されています。
もともと、式(1)では
+0.252603673454155*LipinskiHBD
+0.49581414764576703*NumHBA とHBD、HBAの係数は+なので、HBD、HBAが大きくなると水溶性は高まる事がわかります。
ただ、それだけでは不十分で、
+1.0678749094594537*LipinskiHBD
+0.5755342165654573*NumHBA
-0.45241634258826235*LipinskiHBD*NumHBAとドナー/アクセプター相互作用を導入した方がさらに精度が高くなることがわかります。
Chiパラメータ(χパラメータ)と言うのは、Atomic(Molecular) connectivity indexを表す指標です。
昔はWienerの3歩数とかMCI(Molecular Connectivity Index)などを使ったものでですが、ある程度長距離まで結合をインデックス化したのが、RDKitのChi0-4n,Chi0-4vのパラメータになります。
o-,m-,p-の違いは長距離まで見たChi4nでは差が出ますが、短距離までしか見ないChi0nでは差が出ないという事です。
さらにChi0n*Chi4nまで導入すれば精度が上がる事がわかります。
(細矢治夫先生の、トポロジカル・インデックス(日本評論社)は数学の本と思っている人も多いですが、実にこれは化学の本で、分子のグラフ理論を勉強したい場合には必読の書籍です。)
もう一つ、式(1)と式(2)では重要な違いがあります。
logSが2のあたりの評価です。logSが2と言うことは、100g/100cc水だけ溶けたと言う事です。
普通の測定では、水100ccに100g溶けたところで、実験は打ち切られます。
完全水溶性と言うことです。
式(1)では、完全水溶性の化合物の計算値はlogS=2よりほとんど皆小さくなります。
これが重回帰法の欠点です。自乗誤差を小さくする為には、大きく外れるものを少なくした方が効率が良いので、平均的に悪くしてしまいます。
これをどうにかしようとすると、重回帰法のプログラムからいじらなくてはならなくなります。
つまり、「あるリミットを超えたら誤差を誤差と考えるな」予測値が2を超えたら予測値が3でも4でも自乗誤差は0とする機能を入れる必要があります。
詳しい事はPirikaのこちらを参照してください。
これが、自分はあまりSPSSやRなどの統計解析パッケージを使わない理由です。
数学的に正しい事をしたいのではなく、化学的に正しい解析を行わないと、親水性を表すパラメータを過小評価してしまうからです。
このような式を得ようと思うと、化学の構造式の意味がわかり、識別子の意味がわかるのは大事な資質である事がわかると思います。
そうした事がわからなくても、PLS回帰すれば自動的に組み込まれるので、気にする必要は無い!
そう考える人は、この分野に入って行かない方が良いのです。
LD50
RDKitも完全なものではありません。
どう組み合わせても精度の出ない問題があります。
例えば、LD50の毒性を同じように計算してみましょう。
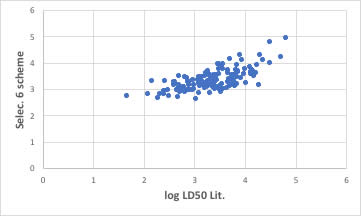
6変数使っても精度は出ません。
そうした時に、計算の合わない化合物を取り出して、例えばHOMOやLUMOの値が大事なのでは無いかと、分子軌道計算の結果を識別子として加えたり、表面電荷の分布値をRDKitから取り出して付け加えたりして精度を高めていくものです。
すでにPubChemの化合物を分子軌道計算したものは、ネットに公開されています。
将来はそうした識別子も含めて全部ネット経由で集められる時代が来たら、お役御免になるかもしれません。
でも当分は、化学がわかり、重回式の意味が理解できる程度にコンピュータがわかれば、後は今回やったように非常に簡単に予測式を立てる事ができます。
logKow, オクタノール/水分配比率
オクタノール/水分配比率は、実はRDKitでも識別子として吐き出してくれます。
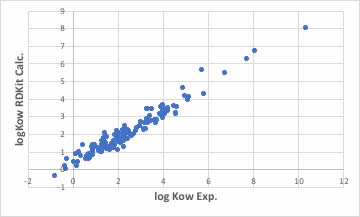
今回使った化合物で計算精度を検証すると上記のようになりました。
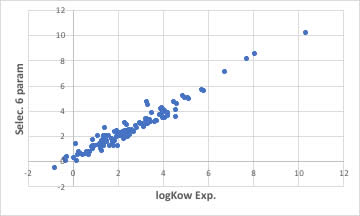
logSと同じように変数選択重回帰法でも6変数で上図のような推算式が構築できます。
RDKitに搭載されているlogKow計算ルーチンと比べて、明らかに精度が高くなっている事がわかると思います。
RDKitのlogKowの計算式を何千個のデータから作ったのかは知りません。
ビッグデータから平均的に精度の高い推算式を作るのと、自分が知りたい範囲に限定して推算式を作るのとの差がここで出ます。
さらに、ベンゼン環についた官能基の位置によって値の変わる識別子を入れた改良式を作ってみてください。
logBCF(生物濃縮性)
logBCF(生物濃縮性)は大まかにはlogKowと相関があります。

ここではデータ数が少ないので正確なことは言えませんが、logKowは分子が大きくなれば、どんどん脂溶性が増して大きくなります。
ところが、生物濃縮性はある分子量までは脂溶性と相関があるのですが、ある大きさ以上になると生体に取り込まれなくなるので、逆に小さくなります。
そこで上に凸の形状になると言われています。
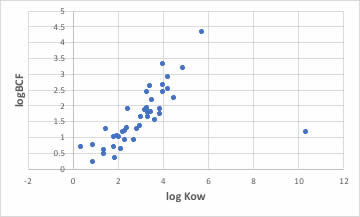
RDKitの吐き出す識別子だけでは(logKowの計算値はないとして)変数選択重回帰式を構築しても、次のような結果にしかなりません。
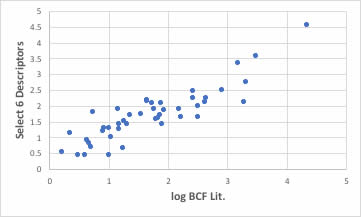
しかし、先ほどlogKowの予測式は作成してあるので、それを含めて重回帰式を構築して見ます。
コンピュータが、logKowに対して上に凸をどう評価したのか見て見ます。
結果は下に示すように大幅に改良される事がわかります。
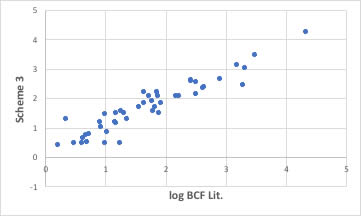
logBCF=-0.00450789115076871*Weight+-0.180534143783582*Chi0n+-0.245838372087646*Chi1n+1.04756743652681*FractionCSP3+0.093508069593571*LabuteASA+0.690310671795674*logKow(Calc)+-0.000055893266885235*Weight*Weight+0.0134054006896844*FractionCSP3*LabuteASA+-1.62787288821536 (3)残念ながら、コンピュータはlogKowを使って上に凸を表現しないで、Weightを使って上に凸を表現しています。
以下の式のようにweightに関して2次関数でWeight*Weightの係数がマイナスになります。
-0.00450789115076871*Weight-0.000055893266885235*Weight*Weight分子量がある点を超えて大きなったら溶解度を下げなさい。
と式に組み込まれている事がわかります。
(2次式の因数分解なので、分子量が幾つなのかは高校生でもわかるでしょう)
このように、RDKitの識別子を使ってある物性値を予測して、それを識別子として加えてさらに新しい物性値を予測する事が簡単にできてしまいます。
自分用のAI-アシスト作成
ここまでできると、JSMEを使って分子のお絵描きをすれば、物性値を返してくれるアプリケーションが作成できます。Pirikaでプログラミング技法](https://www.pirika.com/Education/JP/YMB/Oyako/index.html)を学べば、こんなことも簡単です。 https://www.pirika.com/Program/2019/JSMEwCalc.html
このようにして、自分の欲しい物性値から予測式を作って、より良い物性値になる化合物を探し、実験する。
値が正しくなければ、その値を組み込んで予測式を再構築して行く。
計算機科学の専門家に頼らずに自分ですぐにできてしまえれば非常に実験効率が高くなります。
そこで、マテリアルゲノムやマテリアルズ・インフォマティクスを使うと実験効率が2-10倍上がるなどと言われているわけです。
(JSMEと連動させるにはプログラムの知識が少しだけ必要になりますが、最悪、RDKitのデータを表計算に持っていけば、そちらで計算できるわけですから問題はありません)
大事なポイントは、式の中身をブラックボックスにしない事です。
主成分分析やPLS分析は、コンピュータという数字しか理解できない知能のない機械に化合物Aと化合物Bの違いを認識させるには良い方法です。
知能の無いAIに理解させるために1000を越すビックデータとして分子の指紋(Finger Print)を与える必要があるのです。
しかし、人間には逆に見通しは悪くなります。
一部医薬品のように水に溶けにくいものを何とか溶かしたい場合と再結晶する為にいっぱい溶ける化合物の溶解性、目的が変われば欲しい式の構成も変わります。
たくさんの識別子を使ってブラックボックスで精度の高い予測式が欲しいのか、識別子の意味を考えながら少ない識別子で少し精度が低い式が欲しいのかよく考える必要があるのでしょう。
自動化?
こうした一連の操作の自動化はどうでしょうか?
最初のSmilesのテーブルと物性値を入れれば、自動で推算式を構築してJSMEに組み込む用のプログラムを吐き出すところまではプログラミングの知識があれば簡単に行う事ができます。
そうした時には大多数のデータサイエンティストもプログラマーも必要なくなる時代が来るのかもしれません。
そうなったとしても、化学の知識を持った研究者は、ここで作ったようなソフトを活かしてAIアシストを受けながら生き残って行くと自分は信じています。
このソフト群はMAGICIAN養成講座でも使うので使い方はしっかり押さえておいてください。
今回、RDKitのブラウザーバージョンを公開したのは、これがあればオリジナルのRDKitがいらないからではありません。
オリジナルのRDKitとPythonは是非インストールして使い方を覚えて欲しいのですが、化学の研究者にはちょっと荷が重いのでは無いかと思います。
でも、これで何ができるようになるか、化学者にわかる形で示せば、敷居が少しは下がるのでは無いかと思います。
まだまだ敷居が高くて解らないとか、感想を送っていただければ幸いです。
それでは、Pythonとかプログラミングが使えないと困る事例の一つを、やはりブラウザーでやってみましょう。
ファイル分割
それはSDFファイルの分割です。
pubchemなどから分子の構造と物性値などをダウンロードしてくると、sdfフォーマットでダウンロードされます。
(sdfファイルの詳細はこちらを参照してください。)
RDKitの中にも、化合物の水への溶解性の訓練セットと予測用セットが含まれています。
それを表計算用のソフトにペーストできるように、1行1化合物(物性値などはタブ区切り)になるように整形してみましょう。
すでにPythonとか使って整形できているのであれば読み飛ばしてください。
整形ができて、物性値とRDKitの識別子のテーブルができたら、取り敢えずPLS法で計算してみましょう。
(主成分回帰法で解析した例はこちら)
このページではPLS法計算をブラウザー上で行うことができます。これもPythonとかでPLS計算できるなら読み飛ばしてください。
SMILESの方言
ついでなので、RDKitなど、SMILESの構造式を使う上で問題になる方言をまとめておきます。
SMILES中にこうした記述があると解析失敗になることが多いです。
- 2重結合のcis-, trans- を表すのに、SMILESでは、/, (バックスラッシュ)を使う。\はエクセルなどでは、¥に変換されたりする。SMILES中の¥は半角カタカナのエとなることがある、解析が失敗する。(日本独自の問題なので、解析前に処理するルーチンをつけなくてはならない。もしくはテキストエディターで検索置換)
- 空白があると解析が失敗する可能がある。
- =2= のような不思議な記述(数字は環を切った時に両端に同じ数字をおく。普通は2重結合は切らない。)
- (H)と水素に括弧がついている。水素を入れたい場合には、[H]とカギ括弧にする。
- ニトロ基の表記。正式には、ニトロエタンでは、CC[N+]([O-])=Oのようになるが、CCN(=O)=Oなどと表記されることもある。
- n1cc(nc1)CCNなどのヒスタミン骨格。小文字を使った共鳴構造表記はダメ。
- イソシアニドが、CCN#Cと表記されるSMILESもあるが、正式には、CC[N+]#[C-]にする。
RDKitの解析結果が表示されていなかったら、そこの部分をよく調べてみてください。
情報化学+教育 > 情報化学 > RDKitの使い方
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。