
2018.8.28
情報化学+教育 > MAGICIAN 養成講座 > 講義資料 > 第5回データのクレンジング
MAGICIAN(MAterials Genome/Informatics and Chemo-Informatics Associate Networks)
MAGICIANとは、材料ゲノム(Materials Genome)、材料情報学(Materials Informatics)、情報化学(Chemo-Informatics)を結びつけて(Associate)ネットワーク(Networks)を構築していかれる人財です。
―Liイオンの拡散係数を例にー
マテリアル・ゲノム、マテリアル・インフォマティクスなどの世界で、収集されたデータのクレンジングなどという言葉が出てきます。
最初は意味が解りませんでした。
家庭内にはクレンザー(汚れ落とし)がありますし、お化粧を落とす事をクレンジングなどとも言いますが、このクレンジングは和製英語で、英語には化粧を落とすと言う意味はありません。

英語のCleansing(クレンジング)は洗浄すると言う意味です。
データをクレンジングすると言った場合、英語の本来の意味ではなく、DB中の汚いデータを取り除くと言う和製英語の意味に使っているように思えます。
毒性などの特殊なDBを除き、物性値などのDBは、どういう研究機関の誰が、何時、どういう方法で測定したか、論文などのソースなどが記載されています。
同じ対象であっても様々に異なるデータが記載されていることもあります。
古いデータなどでは単位が異なっていたり、測定条件があやふやだったりします。
データを”クレンジング”してからでなければ、そのままマテリアル・ゲノムで解析というわけにはいきません。
ネット上からLiイオンの拡散のデータが得られたので、これを例題に、データの前処理の方法を説明しておきます。
Liイオンの拡散係数は引火性のない全固体電解質のリチウム2次電池を開発するのに最も重要な物性値です。
Liイオンの拡散データ
たまたまネットを調べていたところ、“リチウム複合酸化物におけるイオン伝導度の系統的評価”京都大学、炭谷晃史 博士論文 2017-03-23 Dkogk04307.pdf という論文が見つかりました。
リチウム複合酸化物628物質のリチウムイオン拡散係数の値が記載されています。
原本のテーブルは容易に表計算ソフトにインポートすることができます。
複合酸化物の、各原子の数はとりあえず手入力で列に入れます(所要時間1時間)。
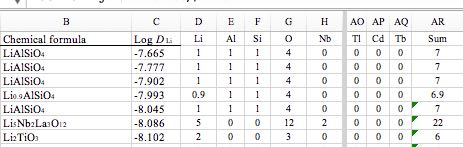
次に組成をパーセント表記にします。
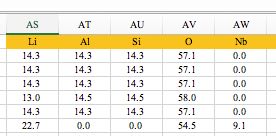
これは、複合酸化物の表記の違いを吸収するために必要です。
つまり、LiAlSiO4とLi2Al2Si2O8は同じものにするためです。
論文中の明らかな誤記、打ち込みミス
Li0.3335Fe0.3335Zn0.3335O5
Li0.05Fe25Zn0.9O4
O5やFe25はあり得ないのですぐわかりますが、容易に修正可能でなければ、とりあえず除外しておきます。
どうすれば、こうした間違いを発見できるでしょうか?
地道に、出来上がったテーブルの列を地道にプロットしてみます。
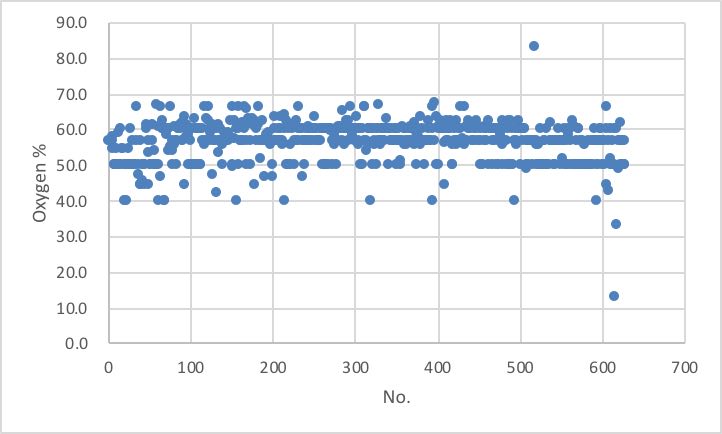
酸素の含有量が33%以下、67%以上は間違いが無いかチェックする事が必要でしょう。
酸素が一番少ない状況は、Li2Oなどで33%, 一番多い状況は、SiO2などで66.6%である事を理解している必要があります。複合酸化物の組成式から原子の列を作る際に生じた入力ミスも、こうしたプロットをしてみると発見される事が多いです。
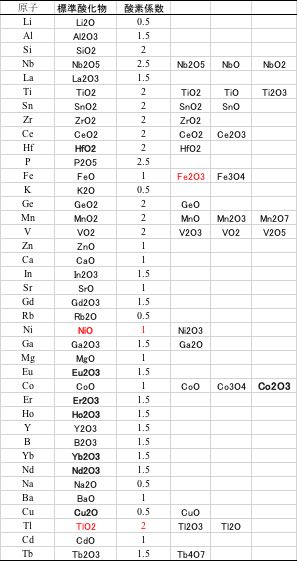
次に、標準酸化物を検討します。
原子のうち遷移金属は酸化数の異なる酸化物が存在します。
その中で、最も一般的な酸化数の酸化物で複合酸化物を作った場合の理想的な酸素含有量を計算します。
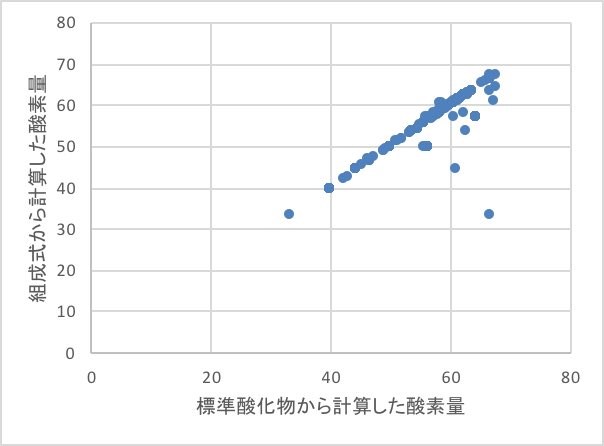
本来は、酸素量は一致しなくてはなりませんが、主に打ち込みミスによって、値が異なるので、テーブルを修正します。
修正が終わっても上図に示すように直線から大きく乖離するデータがあります。これらはほとんどFe化合物でした。
鉄にはFeO, Fe2O3, Fe3O4の形があり、今回はFeOを「標準形」と置きましたが、Fe2O3を原料に用いている場合には、当然、酸化数が異なった複合酸化物になります。
それ以外、微量の酸素欠陥、酸素過剰の複合酸化物も存在することがわかります。
これは、複合酸化物を作る際に、仕込み組成で表示するか、できた複合酸化物の組成を機器分析した結果を表示するかで微妙に変わる可能性があります。
酸化物によっては、融点、沸点が低く調整中に揮散する事もありますし、調整する雰囲気によっても変化する可能性があります。
ここまでクレンジングが終わると、予測式の検討に入ることができる程度のデータセットとなります。
予測式の(予備)検討
作成したテーブルを元に、まず、重回帰解析を行います。
log 拡散係数= 0.015380859375*Li +0.009765625*Al -0.002197265625*Si +0.00390625*O+………
という予測式が求まります。
この重回帰式を求める機能はExcelにも搭載されているのでぜひ試して見てください。
重回帰式は非常に見通しの良い式で、例えば、Siが多くなると、係数がマイナスなので拡散係数は小さくなるとか、Liは係数が非常に大きいだとかがわかります。
ただし、重回帰法は、項目間に相互作用がある場合、現象に非線形性がある場合には予測性能はとても劣るという特徴を持っています。
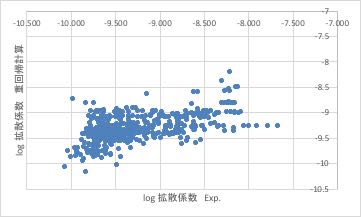
今回のデータ全てをプロットすると上図のようになります。
これでは、ある複合酸化物があった時に、その正しい予測値を得るのは不可能であると言わざるを得ません。
それなのに、なぜ敢えて重回帰計算をするのかというと、もう一つのクレンジングを行うためです。
重回帰計算値でソートをかけてみましょう。
すると、計算値が全く同じになるものが存在することがわかります。
例えばLiAlSiO4という複合酸化物は同じ組成ながら、拡散係数の実験値が-7.665から-9.621と100倍異なるデータが存在することがわかります。
計算値は組成が同じなので同じ値-9.261になる。したがって推算式は精度が出なくて当たり前になります。
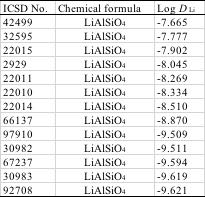
それでは、重回帰計算の結果の-9.261付近のデータを残して、他の値をクレンジングしてしまえば良いのかというとそう単純な話ではありません。
logの拡散係数が-7.665というのは、このテーブルの中で最も拡散係数が高いものです。
それを重回帰計算に合わないからと言って、消去してしまうと本来欲しい高い拡散係数の組成が失われることになります。
テーブルの中には、Li-Al-Si-O系としては、次の10種類の複合酸化物が存在して、データ数は52件存在します。

先ほど作成した、重回帰計算のうち、Li-Al-Si-O系だけを取り出すと下図のようになります。
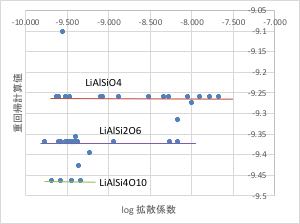
組成が同じであれば、計算値が同じになりますが、その周辺の組成の値と大きく異なる値を残すのは合理的ではありません。
新たに、Li-Al-Si-O系だけで重回帰計算を行い、データのクレンジングを行います。
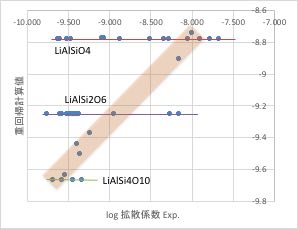
残すデータは、オレンジ色の領域のデータとするのが合理的であると言えるでしょう。
こうした、どのデータを残し、どのデータを削除するのかは、研究者によって差が出る所です。
また、Li-Al-Si-O系のように、いつも合理的にクレンジングできるわけでもありません。
場合によると、クレンジングせずにデータを残して置いて、平均値を使うというという選択肢も大事なセンスになります。
Li-Ti-La-O系では多くの系をクレンジングして消去しましたが、どうしても判断のつかない系は残しました。
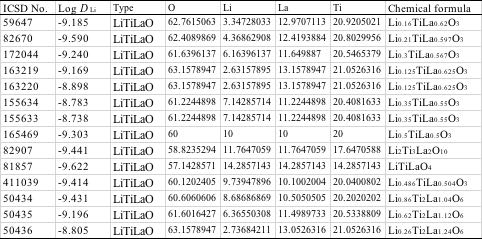
組成が整数比でないものは、結晶系などの違いによって特異的に結果が変わりうるので、無視はできない事もあります。
特に、これらのLi-Ti-La-O系では顕著になるのですが、標準酸化物で計算した酸素量と、実際の酸素量が一致しません。
原子の組成比に加えて、酸素の過剰、欠乏も学習データに加えることにします。 また、特許などを調べると、特異的にTiが好きな会社もあることが分かります。
そこには何か秘密が隠されていることもあるので無視はしません。
今回は、複合酸化物として628系のデータがありましたが、最終的には251系と半分以下のデータを用いて最終的な予測式を作成しました。
このようにビッグデータを使うよりも、質の高いデータを選び出す事の方が成功に繋がるように思えます。
また、クレンジングによって残すデータは、研究者の個性が出るので、この先の結果は大きく異なる事もあります。あくまでも一例として、自分の解析結果と比べてみてみてください。
予測式の作成
ここではニューラルネットワーク(NN)法を用いて推算式を構築します。
重回帰法と比べ、NN法は項目間の相互作用が取り込めたり、非線形性を加味された予測式を作るには適しています。
ただし、できあがった予測式の情報の流れがブラックボックス化してしまい化学的な解析がしにくいのが欠点でもあります。
NN法による物性推算に関しては別の章で詳しく解説しようと思います。
NN法のソフトウエアーに関しては、最近は機械学習のソフトウエアーとして様々なものが流通していますので、適当なものを探して使ってください。学生用のバージョンは所定のサイトからダウンロードして利用してください。
データのクレンジングがうまく行っていれば、比較的簡単に収束して、組成を入れれば予測値を返すシステムが構築できます。
多くのNN法では、中間層のニューロン数の設計が一番難しい点となります。
ネットワークの構造を変えただけでも結果は大きく変化します。
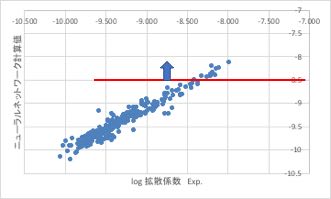
最終的に採用したNNは上図のようなパーフォーマンスを持つものです。
相関係数的にはさらに高いものも作れますが、その分、非線形性が高まり、予測性能が逆に悪くなります。
なれるまでは中間層のニューロンをなるべく減らしたほうが良い結果が得られるでしょう。
NN法で予測値が-8.5より大きくなる(赤線よりも上)構成を押さえておきます。
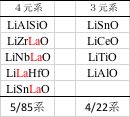
4元系で5種類、3元系で4種類の複合酸化物があることがわかります。
授業で使うニューラルネットワーク法のソフトウエアーはエクセルの表形式でも出力されるので,複合酸化物の組成を入力すれば自動的に予測値を答える計算機をExcel上に構築することができます。酸素の過剰、欠乏は自動的に計算するので入力する必要はありません。
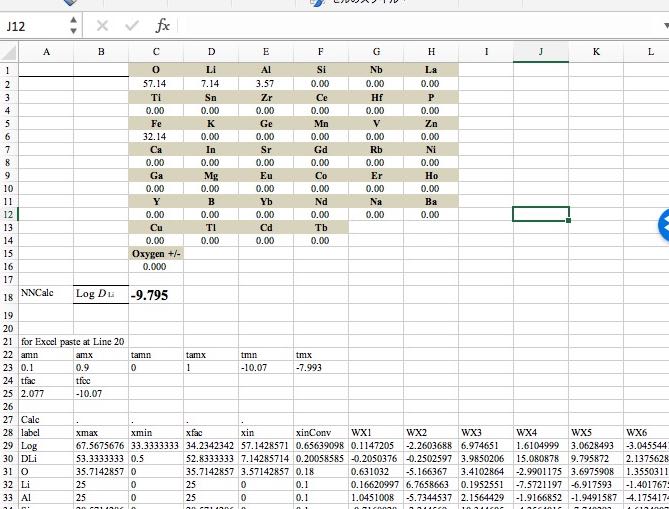
Excelでマクロを書くことができれば,例えばLiの量を変化させた時に,拡散係数がどうなるか,連続計算させることも簡単にできるでしょう。
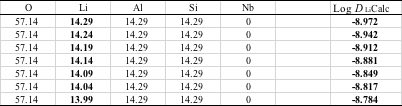
ただし,基本形から大きく乖離する組成を計算することはお勧めしません。
これらの複合酸化物は結晶化ガラスと呼ばれるガラスです。
基本形から大きく外れるものは結晶の構造が大きく変化し予測する拡散係数が大きく外れることもあるからです。
また,予測式はJAVA(C#)のプログラムとしても出力されるので,プログラムが書ける場合には,より高度なアプリケーションを作成することも可能です。 MAGICIAN養成用にはWebアプリの雛形があるので、Weith Matrixをペーストすれば予測値を得るWebアプリが瞬間で作る事ができます。
こうした計算機で,より高い拡散係数となると予測される新たな組成を実験し,結果を予測式にフィードバックし,さらに高い拡散係数となる組成を求めて行く。
予測式は一度作ったら終わりではなく,日々更新して行くものです。
ExcelやJavaの必要な部分だけを差し替え、バージョンを上げていかれる手軽さは非常に重要です。
逆設計
遺伝的アルゴリズムを用いた逆設計については,別の章で詳しく説明します。
ここでは,次の機能を持つJavaScriptのプログラムを作成しました。
- 拡散係数を予測する。
- 連続計算を行う。
- 遺伝的アルゴリズムを使って,基本形の組成を少し動かして,性能を向上させる組成設計を行う
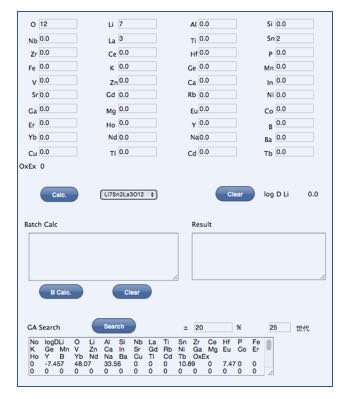
このプログラムは,ブラウザー上で動作するので,PCだけではなく,スマートフォン上でも動作します。
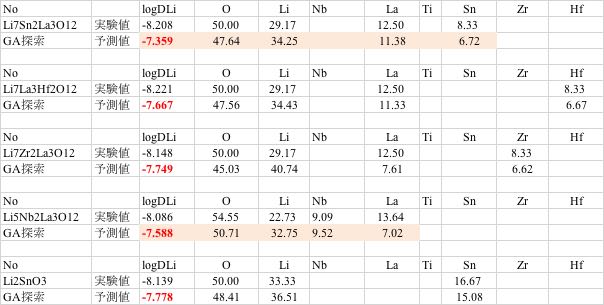
基本組成を入力して,組成を動かす%を入力して探索を行うと改良処方が提示されます。
これは、こうした複合酸化物は大きく組成を動かすと結晶系が変わってしまい推算値の妥当性が低くなるからです。
基本処方は、手入力も可能ですが、NN法で予測値が-8.5より大きくなるものはプルダウンメニューからも選択が可能になっています。
こうして得られた処方を元に、ある原子だけ量を増減して連続計算するルーチンも搭載してあります。
予測式の拡張
今回のデータソースには3元系,4元系の複合酸化物しか含まれていません。
ニューラルネットワーク法などで物性を予測する場合,基本的には外挿性は期待してはいけません。
つまり今回作成したプログラムで,5,6元系の複合酸化物を計算してはいけません。
それでは計算は常に実験の後追いになってしまうので面白みに欠けることになります。
そうした時には,別のソースを当たってみることも重要です。
例えばInterGladというガラスのデータベースがあります。そこには,様々なガラスの組成と物性値が記載されています。
その中で電気伝導度のデータを調べ,電気伝導度の高い組成を抜き出してきます。
こうした組成を中心に予測式の拡張を図るのも一つの手です。
このデータベースには,
Li2S-GeS2-LiI
Li2S-LiI-SiS2
Li2S-P2S5
などの硫化物系の組成も記載されているので,さらに広い範囲に拡張が可能でしょう。
このデータベースを使うのであれば,再び,クレンジングから地道に繰り返す必要があります。
そして,拡散係数と電気伝導度の両面から組成設計を行って行くのがマテリアル・ゲノム、マテリアルズ・インフォマティクスの一番面白いところだと私は思っています。

雑感
LiBの電池は電解質に有機溶媒を用いており引火事故が絶えません。
それに対して、こうした全固体電解質は引火しない次世代のLiBとして、マテリアルズ・インフォマティクスを使って激しい開発競争が繰り返されています。
ネットからデータを拾ってくるだけでも色々楽しめます。
MAGICIANの腕試しにはもってこいなのでチャレンジしてください。
マテリアル・ゲノムを使うと実験などしなくても新しい素材が次々と生まれるような誤解があるようです。
また、スーパーコンピュータや高価なソフト、高額な分析装置が必要であるという誤解があるようです。
そしてそれを使う側にも、高度な数学、統計解析能力が必要であるという誤解があるようです。
その敷居の高さが普及を遅らせているようです。
ニューラルネットワークに化学を教えようとした時に、一番適した教育者は化学者自身です。
本来、化学の知識を元にクレンジングすれば少ないデータ数でも精度は上がるはずです。
それを、中をブラックボックス化して、大量のデータを用いて行うのが現状のマテリアル・ゲノムとなっています。
データサイエンティストに化学を学ばせるのが早いか、化学者がちょっとしたテクニックを覚えて少ないデータで効率的なmaterials genomeを実行するか。どちらが良いかは明らかなように思えます。
その上で、問題に突き当たったら、赤池情報量基準を学んだり、プログラミングの知識を吸収したりしながら、自分の為だけに貢献してくれる、マイAIアシストを作り出していく。
そうすれば当分AIに職を奪われることはないでしょう。
ビッグ・データがある場合には、統計的にみれば、正しい答えをAIは答えてくれるという間違った認識があります。しかし、それは間違いです。
最近読んだ新井先生の著書に簡単な例でその事が記載されていた。
「私は、山口と岡山に行った。」
この文章を読んだ時に、人間であっても、山口が土地の名前なのか人の名前なのかはわかりません。
前後の文脈を理解する能力が必要になります。
ネット上の全ての文章の、ビッグデータを解析したところで、山口が土地の名前か、人の名前かは確定できません。
“この山口は人名”なのでクレンジングしようと思えるのは、人間だけで、その人の自己責任でクレンジングした結果、本来掘り下げるべきデータを除外して大発見を逃す事も十分あり得ます。
今回の例はあくまでも素人の筆者がクレンジングした結果なので、結果が異なっていても気にする必要は全くありません。
様々な立場の研究者とモデルの違いを議論し、可能であれば実験結果、論文と、特許等からのデータ値を加味し、再構築して研究を加速して行くのが大事なのだと思います。
情報化学+教育 > MAGICIAN 養成講座 > 講義資料 > 第5回データのクレンジング
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。