
2021.04.25
情報化学+教育 > MAGICIAN 養成講座 > 講義資料 > 第9回 新しい解析ツール
MAGICIAN(MAterials Genome/Informatics and Chemo-Informatics Associate Networks)
MAGICIANとは、材料ゲノム(Materials Genome)、材料情報学(Materials Informatics)、情報化学(Chemo-Informatics)を結びつけて(Associate)ネットワーク(Networks)を構築していかれる人財です。
2020年の横浜国大の授業は全てZOOMになりました。
ZOOMの良いところは、時間や距離の制約がなくなることでしょう。
私の授業は春学期なので、スタートが遅れた事はありましたが、8月には終了しました。
まだ学び足りないという学生と始めた課外授業=MAGICIAN養成講座も19回を超えました。
途中から、東京の社会人研究者1名、名古屋の社会人研究者2名、大阪の社会人研究者1名と毎週土曜日の午前中10時半から12時までみっちり講義をしています。
時には英語で講義したり、海外の方が参加しても大丈夫です。
文字通り、時間や距離を超えて人的 NetworkがAssociate し始めてきました。
学生が開発してきた、解析ツールがいい味を出してきました。
養成講座に参加している方と名前を考えて”GROVE”という名前にしました。

GROVE Gene-based Regression Offering Valuable Equations.
貴重な式を提供する遺伝子ベースの回帰分析
っていう意味になります。
もちろん、開発した学生の名前が林なので、先にGROVEが出てきて、皆んなであれこれ英語を当てはめました。
MAGICIAN用の解析ツールには、重回帰分析、主成分解析(PCA), PLS、ニューラルネットワーク法など様々なものがあるのに、何故、今更と思われるかもしれません。
昔の誤差論。(▶︎をクリックして開く)
私は、2007.2.6に東大の舩津先生の所で行われた、俯瞰環境工学寄附講座の公開シンポジウムで、「物性推算と逆設計 構造から物性へ-物性から構造へ」というタイトルで発表しています。
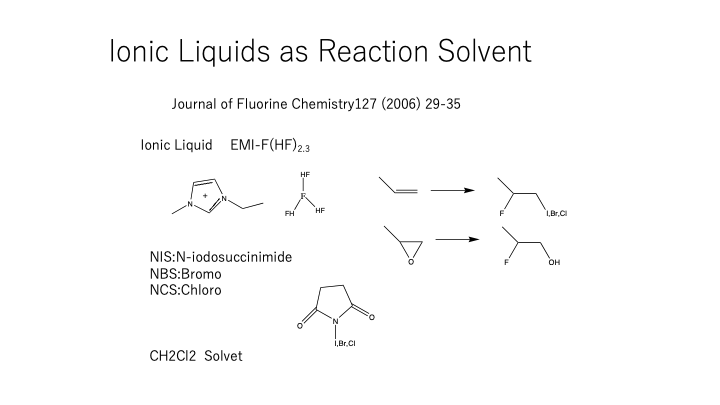
題材はイオン液体です。
全部では40ページもあるので、引用は一部にしておきます。
2007年の話なので割り引いて読んでください。
データ解析にはいろいろな方法がありますが、それぞれ特徴があります。
まとめるとこのようになります。
データ量や求める精度によって使い分けないといけにという話です。
この発表では、新たに開発したMIRAI法(当時はPEM法と呼んでいた)を使うという話です。この方法はこちらで説明しています。

どのような解析に使うのかというと、イオン液体中でのフッ素化反応です。
なんでも良いですけど、今回はイオン液体への応用の話でしたので。
論文には様々な基質の収率が記載されていました。
そのデータを元にまずテーブルを作成します。
欲しい目的変数(収率)を説明する説明変数の組みを定めていきます。
ここでは、分子軌道計算した時のHOMO,LUMOや電荷などを利用しています。
その辺りは、今でも大きくは変わっていないでしょう。
せいぜい、私の計算がMOPAC程度だったものが、ガウシアンへ変わる程度でしょうか。
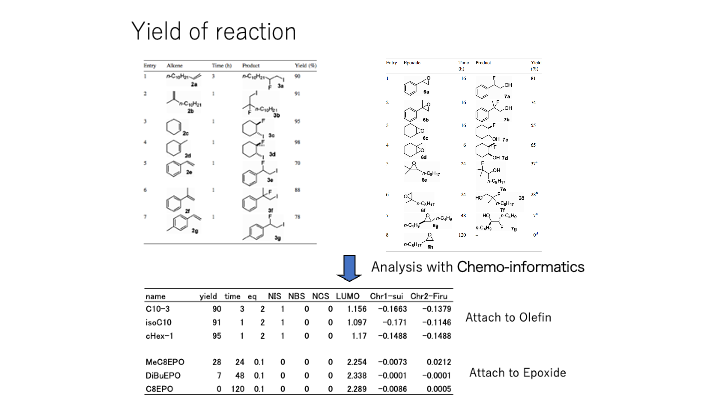
そしてモデル式を作成していきます。
MIRAI法では図の下にあるようにPower関数の掛け算で物性を予測します。
PEMの名前の由来はともかく、単にフィードフォワード・タイプのニューラルネットワーク法の1種だと思えば間違いありません。
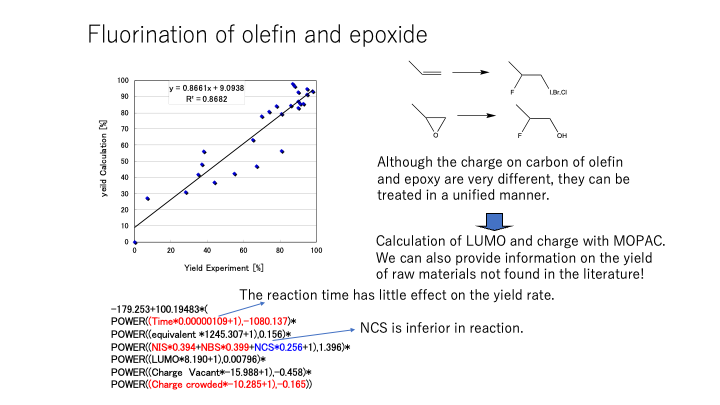
実にこの部分が言いたいところです。
化学者の常識が大事なのです。
AIには常識が無いのです。
逐次反応でないなら、120分で収率0%なら5分でも30分でも収率は0%と化学者にはわかります。
でもAIにはきちんと教えないとわからないのです。
収率は100%を超えることはないし、0%より小さいこともありません。
相関係数だけで見ていてはダメなのです。

その際にどのような解析ツールを使っていようと、大事になってくるのが、教師データと計算値の外れ方をどう評価するかです。
通常は、(というか私はそれ以外を見たことがないのですが)二乗誤差(自乗誤差)を最小にするように動作させます。
誤差逆伝播法であろうが、フィードフォワード法でもそれは同じです。
ところが自作のソフトであれば、色々な誤差の取り方を試すことができます。
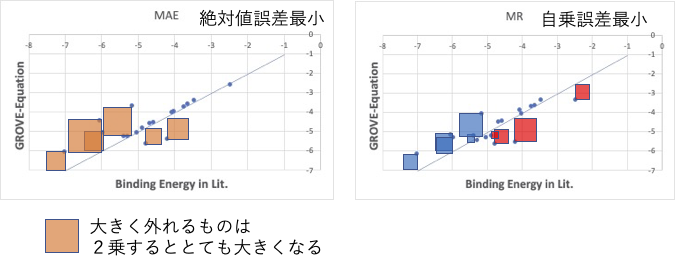
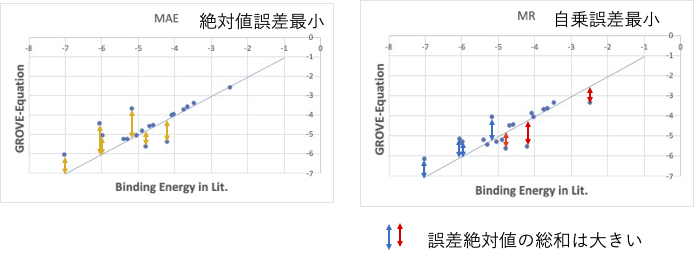
例えば、誤差の絶対値を最小にするように動作させると、大きく外れるものを分離することができます。二乗誤差を最小だと、大きく外れるものは2乗するとさらに大きな誤差になり、修正圧力が高くなります。そこで化学系のように元々の教師データが汚いデータだった場合に誤差の絶対値最小法を使うと汚いデータが浮き上がって、修正しやすくなります。
%誤差だと値が小さいものほど修正圧力が高くなります。
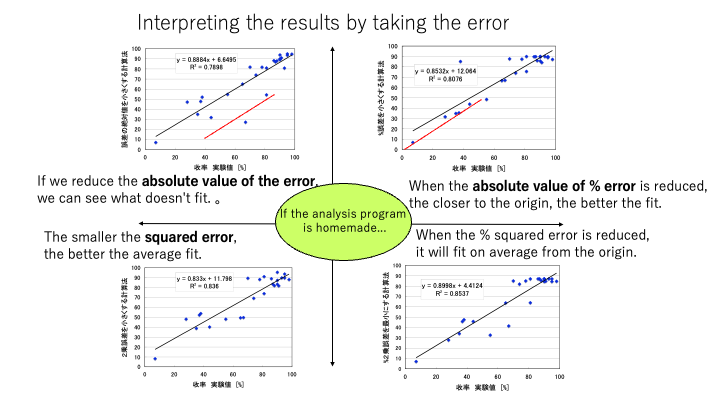
そのように誤差の取り方を変えて推算式を構築します。そしてその式の中の時間に関わる項を抜き出して評価すると次のようになります。
反応時間が長くなった時にどうなるかの評価が、誤差の取り方で全く変わってくるのです。
どれが正しいのかよく聞かれます。
それがわかるのは、“実際に研究している化学者だけ”というのが私の発表の概略でした。
発表のタイトルは「物性推算と逆設計 構造から物性へ-物性から構造へ」です。
物性ではなく、ここはプロセスですが、最適プロセスを逆設計しようとした時に、グラフ自体は大きく変わらないのに、時間の効果が誤差の取り方によって全く変わってしまいます。
これが、AIを用いた物性推算・逆設計の多くが失敗する原因になっています。
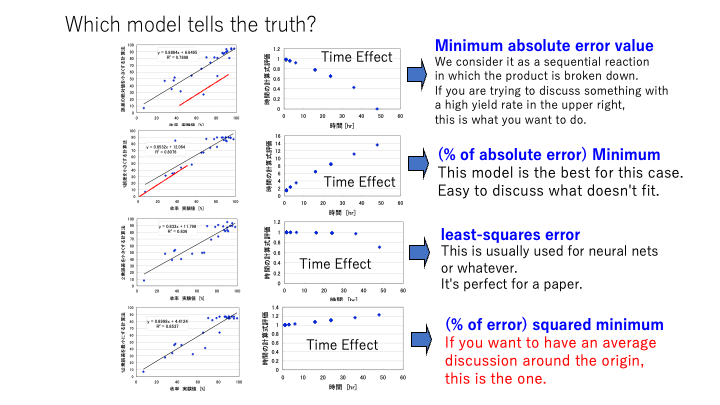
化学者は情報化学を毛嫌いしないでと訴えたわけですが、まー、時代がまだ追いついていなかったのでしょうか。
逆に自分がドンキホーテになって実験化学の水車に立ち向かったけど無駄だったような気がします。

まー、そんなワケで私以外誰も使わない解析方法でした。
たまたま、MAGICIAN養成講座を受けている生徒に、「データセットの中の値の一つをめちゃくちゃな値を入れました。どれが怪しいか解析してください。」という課題を出しました。

これは、データ・クレンジングの基礎になります。
データをスキャナーでスキャンして、OCR(光学文字認識)にかけたりすると、ある確率で8と3を誤認識することがあります。Sと5は文字と数字なのですぐわかりますが、7と9を間違って読み込んで学習データにしてしまうことは良くあります。
ニューラルネットワーク法のように非常にフィティング能力が高いと、教師データがどんな値(例え間違った値)であっても収束してしまいます。詳しくは、こちらのページで確認してください。この例では、CH2(OH)CH(OH)CH3という化合物。データベースには35.5dyn/cmという値と72dyn/cmというデータがあります。
どちらもかなり信頼のおけるデータベースです。
これをニューラルネットワーク法で推算式を構築すると、どちらの値であっても収束してしまいます。
これは非常に困った性質です。
私がわざと間違えて入力したデータを解析者はデータ・クレンジングできない事になってしまいます。
データ数が少ない時にはそれでも統計諸量を見ればわかるかもしれません。
でもデータ数が増えたらもうダメです。
そうした時に、誤差の取り方を変えると、「線に乗るものはどんどん線に乗せて、外れるものはどんどん外れるように動作させることができる方法」を学生に教えたところ、一人の学生が頑張ってプログラム化してくれました。
それがGROVEです。
GROVEの解析例
それでは1つだけGROVEの解析例を見ていきましょう。
去年(2020年)の4月ごろ、お茶の成分(カテキン)がCOVID-19スパイクの侵入と融合を阻害すると言うニュースがありました。奈良県立医科大学が新聞発表しているので、ご存知の方も多いかもしれません。
それに関して、
Identi ficaion of Dietary Molecules as Therapeutic Agents to Combat COVIDー19 Using Molecular Docking Studies:Mohannad Faheem Khan,et al
と言う論文で、植物由来の成分とCOVID-19ウイルスのスパイクとの相互作用を、Molecular Dockingで調べているものが見つかりました。
ざっくり言ってしまえば、ポリフェノールなどの抗酸化作用を持つ化合物は、COVID-19スパイクの侵入と融合を阻害しやすいようです。
次のデータを表計算ソフトにコピペしておきましょう。
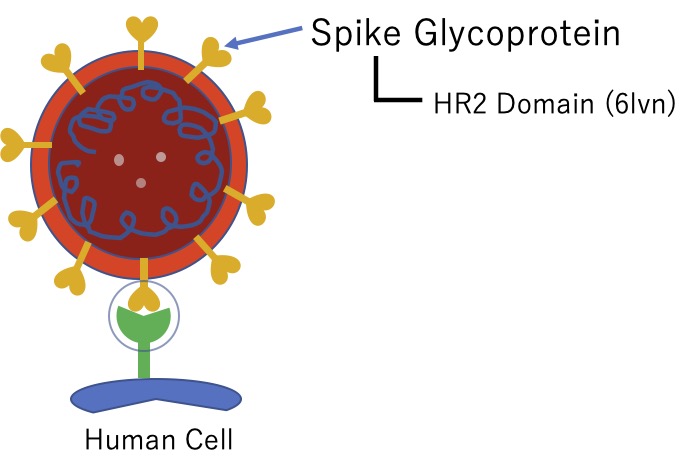
6lu7: activityagainst main protease
6lvn: HR2 domain of spike glycoprotein
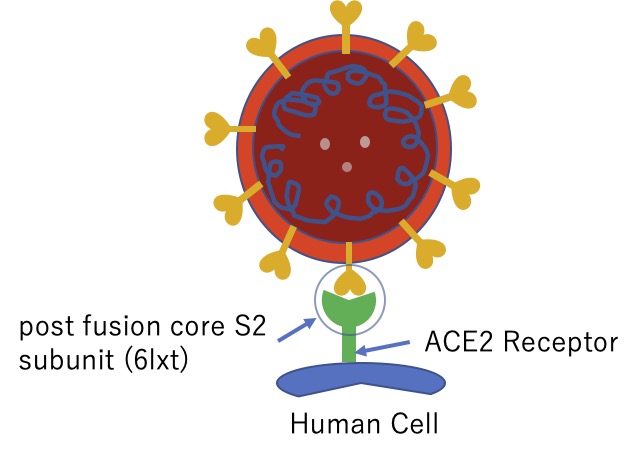
6lxt: post fusion core S2 subunit
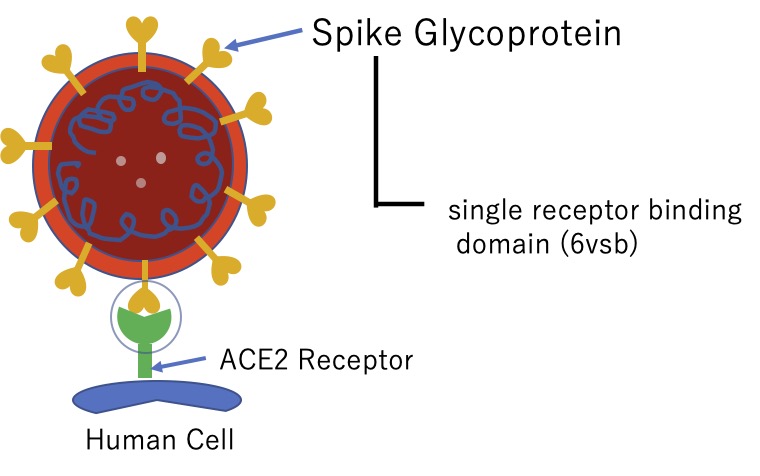
6vsb: single receptor binding domain of spike glycoprotein
binding energyの単位は kcal/moleです。
最も効果の高いものは、EGCG(epigallocatechin gallate)だと言っています。
ちなみに、Remdesvir やトランプ大統領が使って有名になった、Chloroquineなどのbinding energyも記載されています。
[カテキン類の精製方法リンク切れMOOC]はこちらを参照してください。
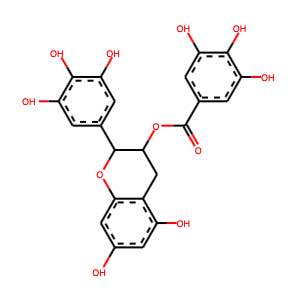
各成分の構造式はSmilesの形で付け加えておきました。
そのSmilesの構造式からHansen Solubility Parameters (HSP)を計算してbinding energyを予測する事にします。
ただし、ここで使うHSP(ハンセン溶解度パラメータ)の値は、YMB-2019のパラメータを使って計算したものです。 HSPiPにはまだ搭載されていません。
YMBというのはYamamoto Molecular Breakの略で、私たちが市販しているHSPiPというソフトの物性推算を行う部分の名称です。
HSPとbinding energyを載せておきます。
やりたいことは、4つのbinding energyをHSPを用いてモデリングする。そして予測化合物のbinding energyを見積もる事です。
予測化合物は、たまたまLinkedInの知り合いがコメントしていた、ファイザー社の化合物、PF-07321332という化合物です。
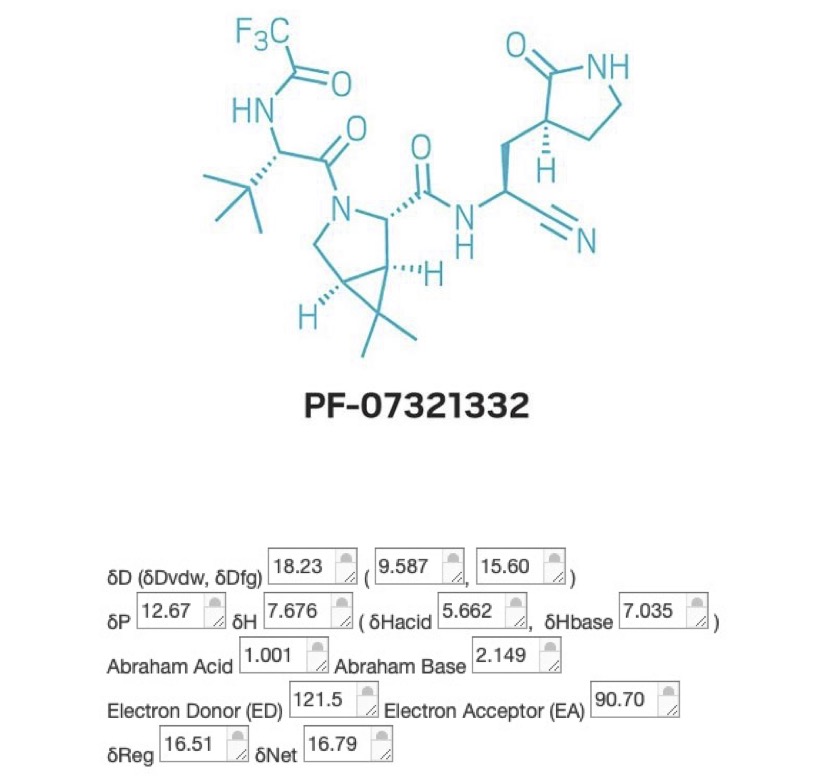
このファイザーの化合物を見ていて、よく似た化合物を最近計算したのを思いましました。
最近化粧品で防シワ効果が話題になっている、ポーラ化成のNEI-L1。末端がカルボキシル基なのでdHが少し高いのですが記憶どおり、ほとんどファイザーの化合物と同じHSPになります。(エステル化すればほとんど同じ?)
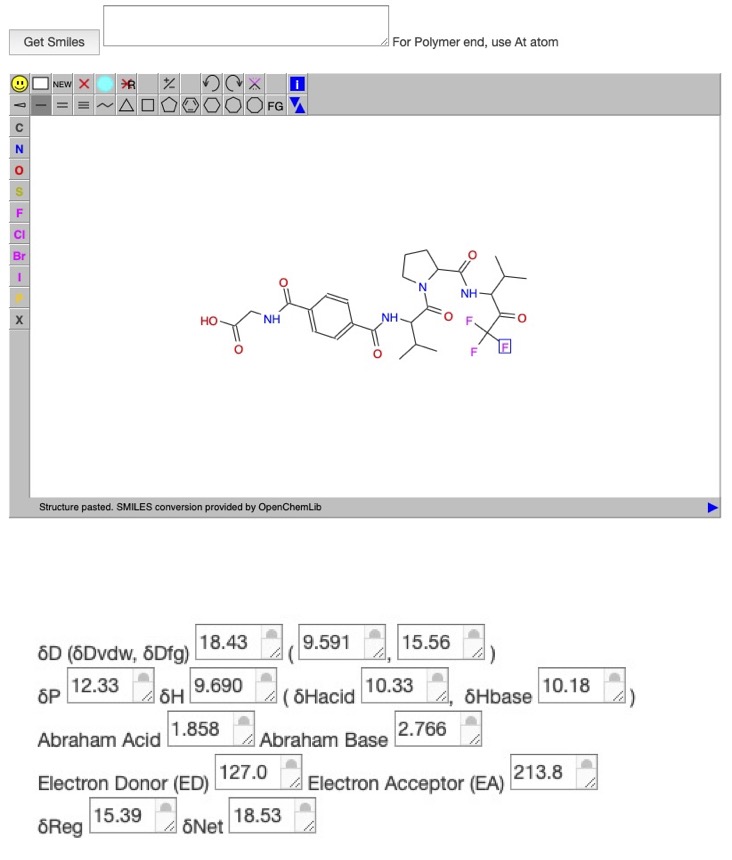
この2つの化合物のbinding energyを予測する事にします。
GROVE解析結果
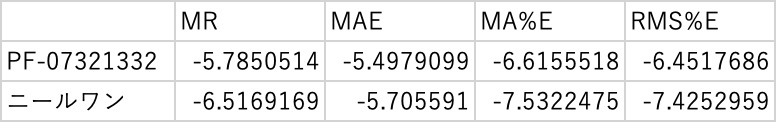
Main protease、6lu7に対するbinding energy
まずは、メイン・プロテアーゼ, 6ln7のbinding energyをHSPを使って予測することを考えます。
Big Data があるならともかく、そんなものは無いので、通常の重回帰法で式を作ります。
通常の重回帰法はExcelの分析にもその方法が入っているくらいで、簡単に式が求まります。
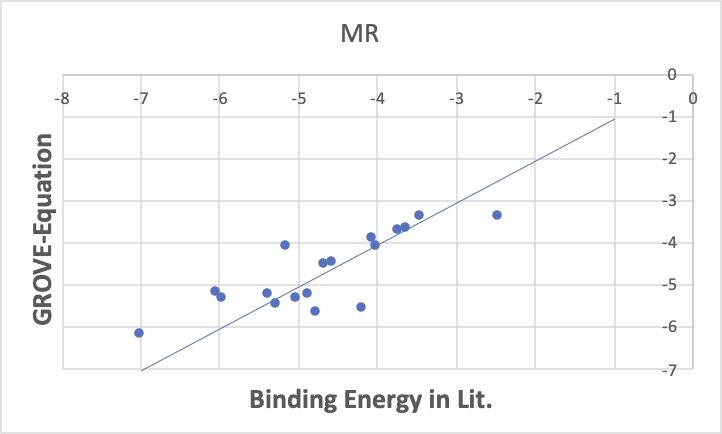
式の特徴は、Y=Xの線の上に平均してばらつきます。
この時の、線への距離の二乗の総和が一番小さくなるように、各HSPへの係数が求まります。
binding energy=-4.8617*dD16vdw-3.7375*dD16fg+4.3207*dD-0.02636*dP16-0.09217*dHAcid16+0.1857dHBase16*H2+0.0068821*MVol14nov-8.0296*Ovality14nov+33.24507
となります。この式が得られれば、あとは任意の化合物のbinding energyは化合物をYMBで計算するだけで計算することができます。
ここで問題にしたいのが、誤差の取り方です。
おそらく、このbinding energyはMolecular Dynamicsか何かの方法で、メイン・プロテアーゼ, 6ln7との相互作用を計算して算出したものです。分子をひっくり返したり、動かしたり、水和させたりすれば値が大きく変わる汚いデータでは無いかと思います。それを重回帰法のように”大きな誤差を減らす”ような圧力のある方法で評価してしまうと、どれが汚いデータなのかがわからなくなります。
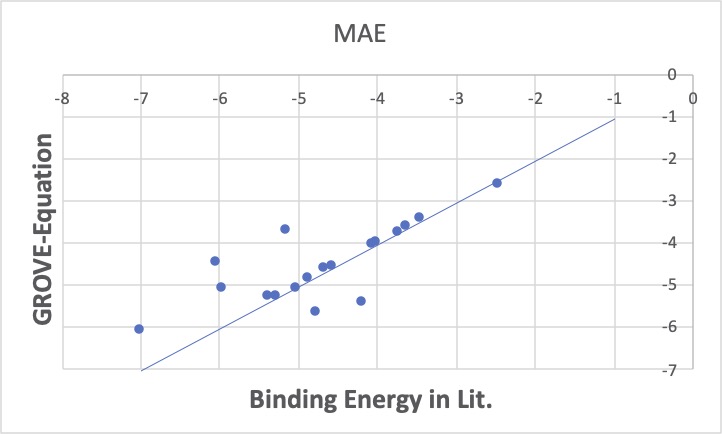
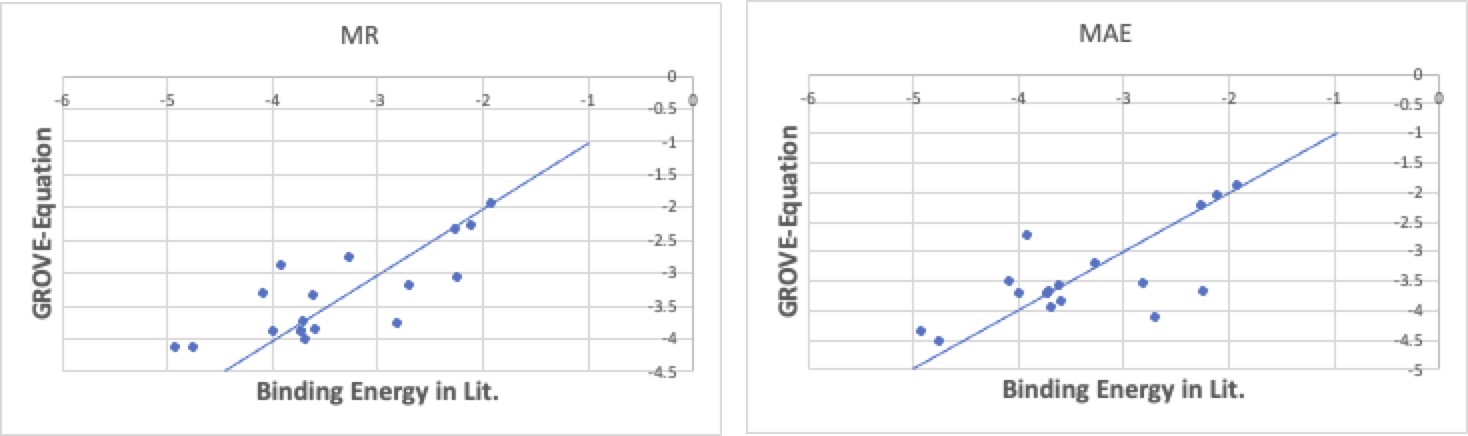
そのような時に、GROVEで、MAEを指定してモデル式を作成します。
すると、ほとんどのものは直線の上に乗り、5つが線から遊離します。
この方法の特徴は、誤差の絶対値の総和を最小にするように動作するため、線分の上に来る外れたものと、下にくる外れたものはバランスが取れている必要はないので、このように線分の上に来るものの方が数も多く、誤差も大きくなります。
また、このような答えの得られる理由のもう一つは、誤差逆伝播法などとは異なり、フィードフォワード法をとっている事にあります。誤差逆伝播法は、教師データが正しいとい前提に立っています。そこで計算値との差が大きいのは、計算値が悪いという前提で処理されます。
ところが、化学ではそう捉えるのは危険です。 測定ミスなんていくらでもあります。
そこで、「合わないものは外れても良いから、合うものはよく線の上に乗るような重回帰係数を求める」のがこの方法の特徴です。
この結果だけから、どちらが正しいモデルであるか答えるのは難しいです。
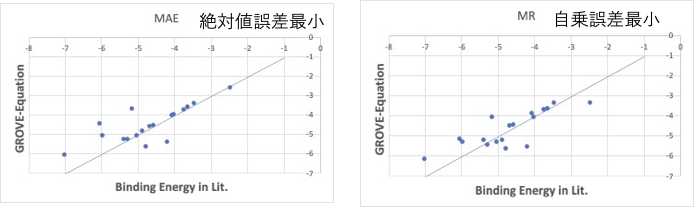
一つ言えることは、解析ツールとして最小二乗法のプログラムしか持っていないとどちらが良いかの議論もできないということです。
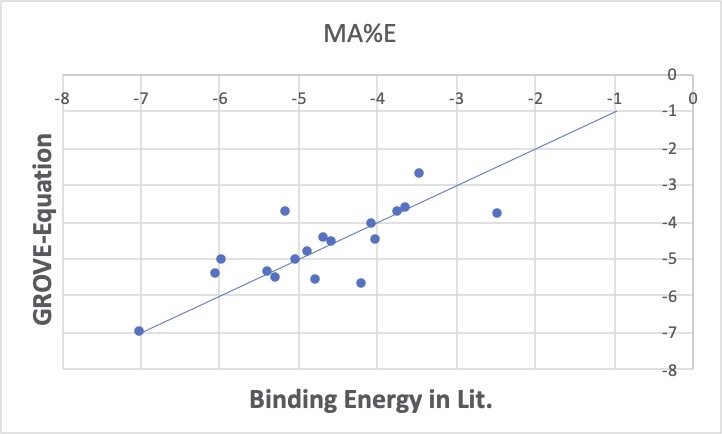
さて、なるべくbinding energyの小さいもの(マイナスの値が大きいもの)をモノを探したいのがここでの課題です。MAEで求めたものはどちらかというと値が大きいものの方が直線に乗ってきています。その時にはMA%Eオプションを使います。
この方法の特徴は、ある領域の精度をあげたい場合に使います。
この場合は、binding energyの小さいもの程よく線に乗るような重回帰係数を求めます。新規の薬を設計して、その薬の性能に自信があるのなら、既存のものよりもっと良くないか?調べたくなります。その時には、binding energyの小さいところで性能の良い係数を使って評価するのが合理的です。
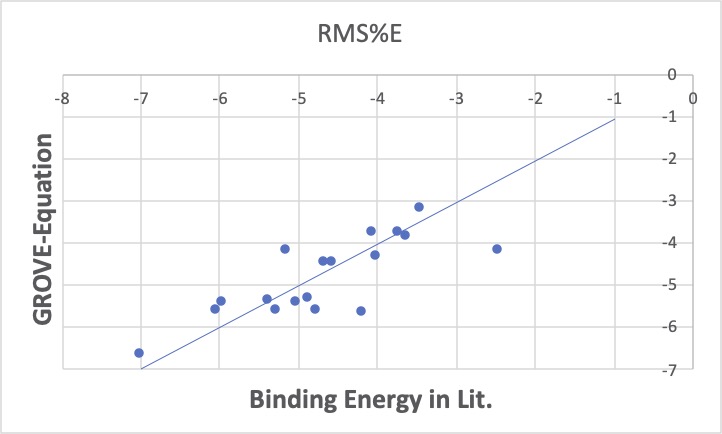
RMSEオプションは、理想的に働けば、通常のMultiple Reguressionと同じ答えを返します。
ただし、フィードフォワード法なので少し異なります。
RMS%Eオプションは、汚いデータあまりないという前提で、ある領域の精度を平均的に高くなるような重回帰係数を求めます。
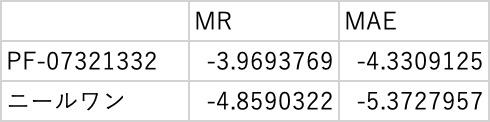
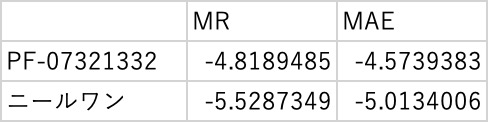
方法論による予測化合物のbinding energyの違いは次のようになります。
どの重回帰係数を用いた場合でも、ファイザーの化合物、ニールワンは大きなbinding energyとなることが予測されます。
元々、ファイザーの化合物はコロナ用に開発されているので、値が大きく予測されることは当たり前かもしれません。
しかし、ニールワンは化粧品で、しかもシワ改善用の化合物です。
鼻の周りにこの化粧品をつけると、シワも改善されて、コロナにかかりにくくなるなら非常に面白いですね。
HR2 domain of spike glycoprotein、6lvnに対するbinding energy
post fusion core S2 subunit 、6lxtに対するbinding energy
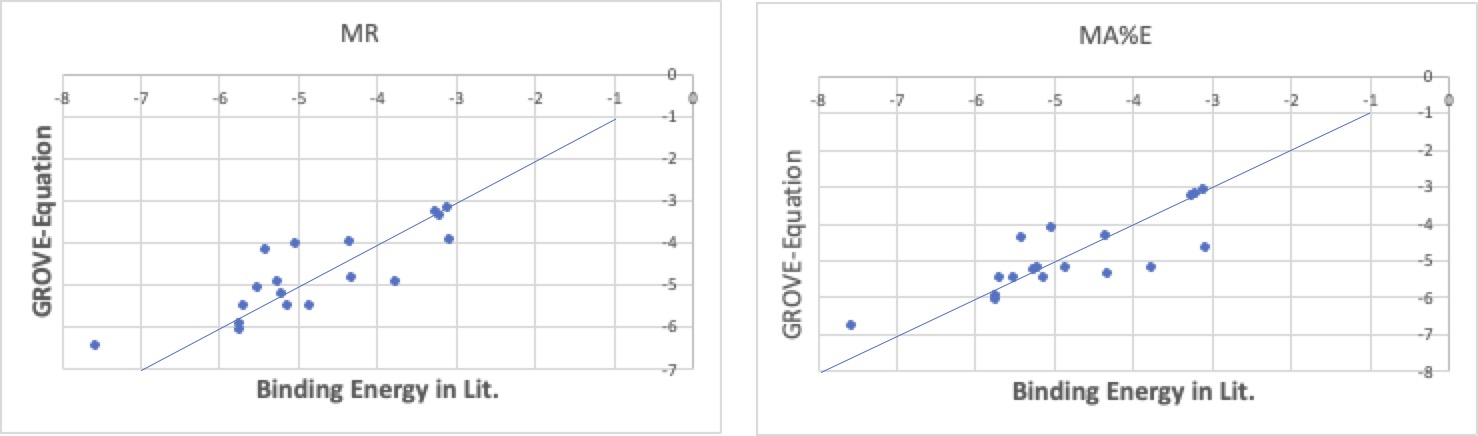
single receptor binding domain of spike glycoprotein、6vsb:に対するbinding energy
この場合には、元々MRでも精度が高く方法による違いは余りありません。
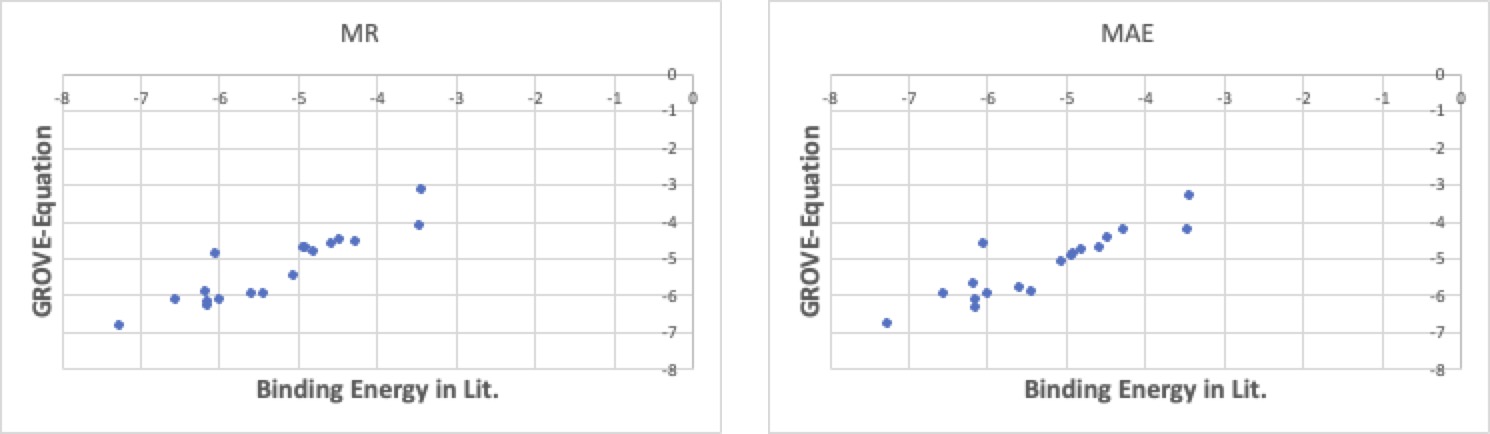
このように、
GROVE Gene-based Regression Offering Valuable Equations.
を使うと、式の形は、通常の線型方程式なのですが、係数の意味が大きく異なってきます。
化合物の場合には、どれか一つのカラムの値を変えるような分子設計はできません。
しかし、触媒設計などでは逆設計を行う際に、こうした逆設計用の解析ソフトはなくてはならないツールだと言えます。
GROVEは現在のところ、MAGICIAN養成講座への参加者限定で利用できます。
すでに、ver.2.8 ver.4.1までバージョンアップしています。
開発している学生は、私の「学生は死なない程度の失敗を全部やり尽くして社会に出る」という方針に従って、苦しんでいます。
まー、バイトの時間を開発に充てられるように、開発ソフトのスポンサーになってくれる企業を募集中というところですか。
以上のように、
「物性推算と逆設計 構造から物性へ-物性から構造へ」と言ったときに、どんな式を立てたときに、どのような特徴がある式なのかは、逆設計の時に大きく効いてきます。
重回帰法や、PLS, PCA, ニューラルネットワーク法という前に、「誤差とはなんぞや?」に関して良く考えるのは大事な事だと思います。
ポーラ化成のニールワンをコロナに試してみてくれる人はいませんかね???
情報化学+教育 > MAGICIAN 養成講座 > 講義資料 > 第9回 新しい解析ツール
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。