
2013.4.2(2020.12.1改訂)
その他の化学トップページ
分子構築用ソフト、Y-Molの使い方
化学工学の学生にとっては、非経験的分子軌道法であるガウシアンなどは費用対効果(学生だから費用と言うよりは学習にかけられるエネルギーというべきか)が低いのでMOPAC程度ができればいいと書いた。MOPACの計算環境の構築については別ページで説明したのでそちらを参照してもらうことにして、分子の組立について説明しよう。
分子軌道法を用いた計算値を得たい場合には、まず最初の関門は、分子の3次元座標を得ることだ。炭素は手が4本あり、水素は手が1本とかいう化学の基本的知識は必須だ。炭素は2重結合を作った場合には手が3本になり、3重結合を作った場合には手が2本になる。そうしたときには3次元の構造として
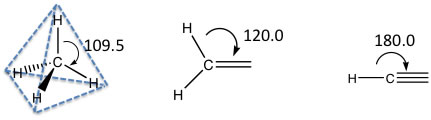
のように、ピラミッド(4面体)構造、平面構造、直線構造を取ることが理解できていなければ、話にならない。この場合の炭素をSP3、SP2、SP炭素と呼ぶ。これは例えばSP3の場合、S軌道と3つのP軌道が混成をおこして、4つの等価な軌道を作っているという言い方になる。
酸素は手を2本しか持っていないので、アセチレンのように直線化合物になると思うかもしれないが、酸素にはローンペアが2つあるのでSP3のピラミッドに近い構造をとり、4つの頂点のうち2つには原子がいない状態になる。それを全て説明しているときりがないので、Y-Molを使った分子の組立に取り掛かろう。(最悪、それらを理解せずに適当に分子を組み立てても、よほどひどい入力構造でなければMOPACで構造を計算すれば正しい構造にたどり着くだろう)単純には手の数からおおよその構造を作ることが出来る。
Y-MolはHTML5+CSS+JavaScriptを使って開発している。
Y-Mol13a (2013.4.2)
ブラウザーの判別で使えるブラウザーであったら、上のリンクをクリックし、別画面でプログラムを走らせながら説明を読み進めて欲しい。またY-MolはYNUSのサブセット版なので詳しい説明に関してはYNUSの使い方を参照にして欲しい。
正しいブラウザーを使っていれば下のような画面が表示される。
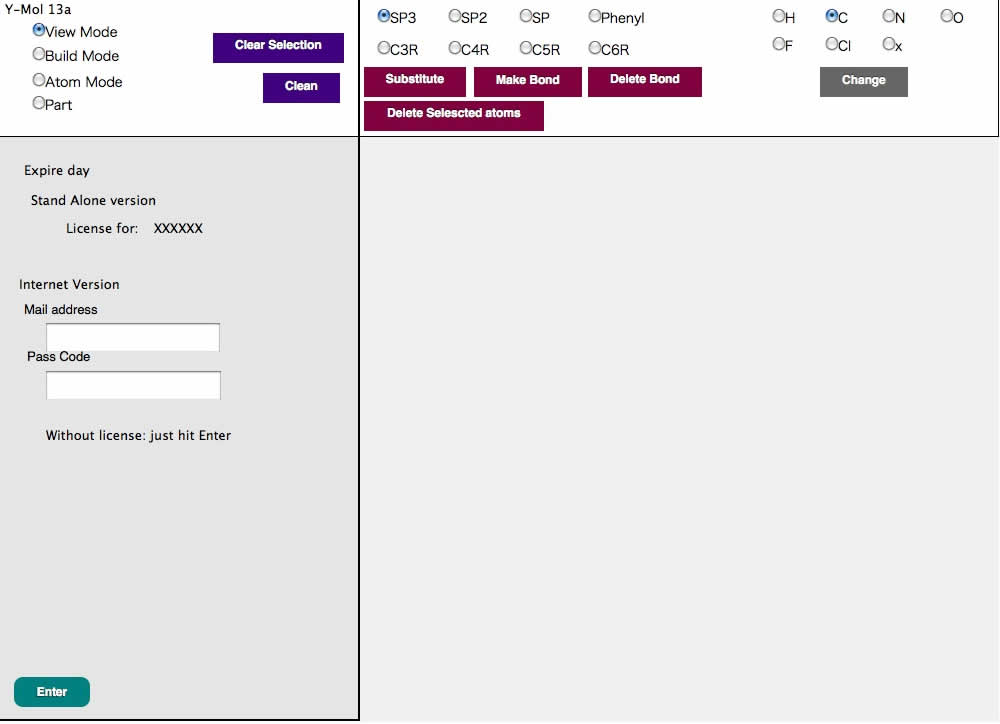
左下のEnterをクリックするとY-Molがスタートする。(スタンドアローン版は授業の時にダウンロードサイトを教えるので聞き逃さないように)
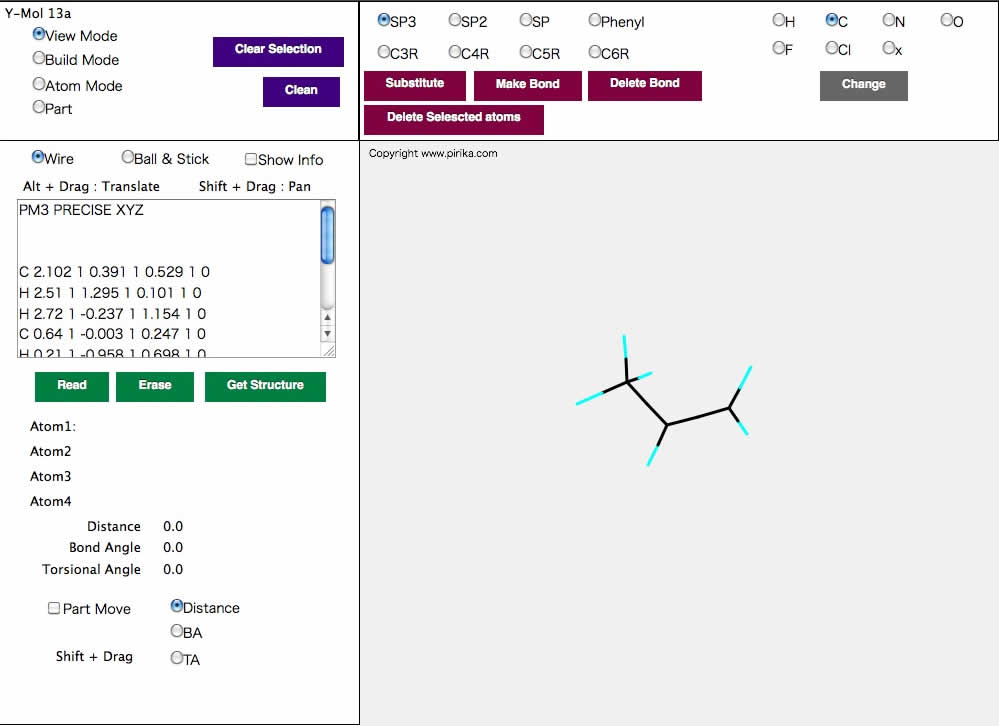
構造を入力するテキストエリアには最初はプロピレンの構造が入っているので、Readボタンを押してみよう。右側のキャンバスに分子が表示される。キャンバスでマウスをドラッグ(マウスボタンを押しながら動かす)すると分子が回転する。Altキー(MacではOptionキー)を押しながらマウスを左右にドラッグすると分子が移動、Shiftキーを押しながらドラッグすると拡大、縮小される。

左上のラジオボタンでジョブを指定する。例えばBuild Modeを指定すると、置換、結合の生成、消去、原子の消去の操作を行うことができる。(上の真中のコマンドが有効になる)ViewMode以外のModeではキャンバス中の分子の原子をマウスでクリックすると赤く表示される。
操作の基本は元になる原子を先にクリックして、操作する原子を次にクリックすることだ。
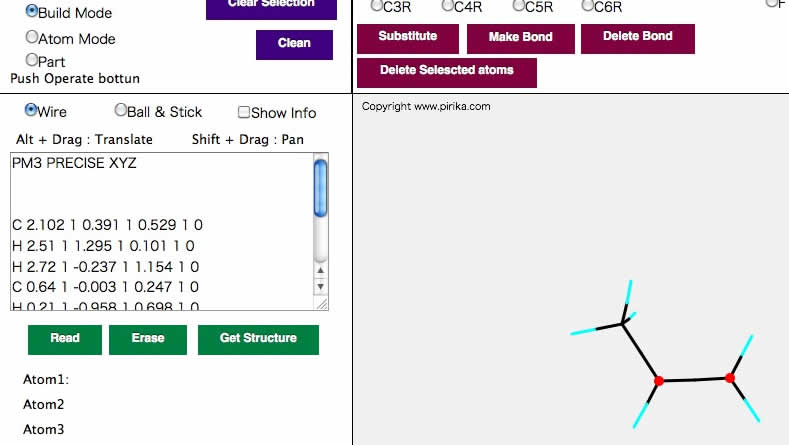
例えば上の図のように2つの原子を指定してみよう。 そしてMake Bondボタンをクリックする。
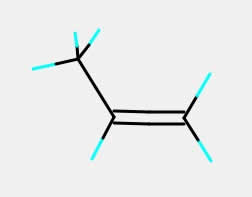
指定した原子間に結合が生成され、2重結合になる。
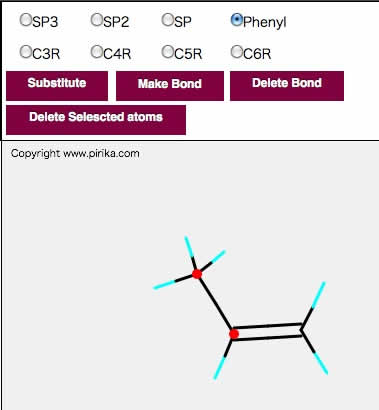
このように先に2重結合の炭素を指定して次にメチルの炭素を指定して、Phenylを選択してからSubstituteボタンを押すとスチレンの構造になる。
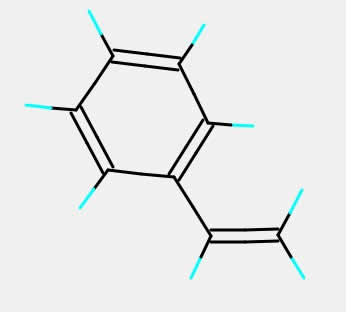
このように2番めに選択する原子は、それに付加している原子は(この場合は3つの水素)皆消去され、置換基に置き換わる。しかし、例えばベンゼン環の炭素を2つ指定して置換基操作を行なっても、消去する原子を辿って行くと元になる原子が出てくるので、置換基操作は行われない。
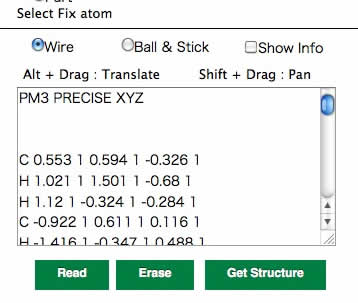
テキストエリアで、Ctrl+A(全選択、Macではcommand+A)し、Ctrl-C(MacではCommand+C)して、メモ帳などに貼り付ける。

Y-Molでは結合情報も出力されるが、MOPACの計算には必要ないので、19行目以降を消去してStylene.datというファイル名でセーブする。
そしてWinMopacを立ち上げ、(走らせ方はMOPACのページを参照)先ほどのファイルを指定する。
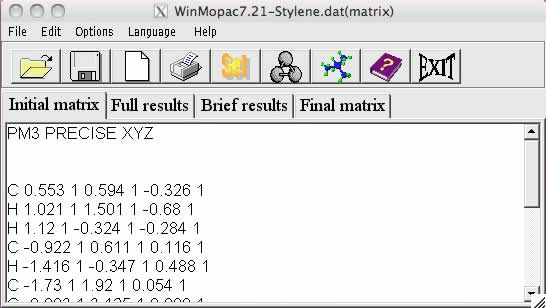
Initial Matrixに構造が読み込まれ、MOPACを計算することが出来る。
最初の構造と、MOPACで構造最適化した後の構造、どちらがどちらだか判るだろうか?
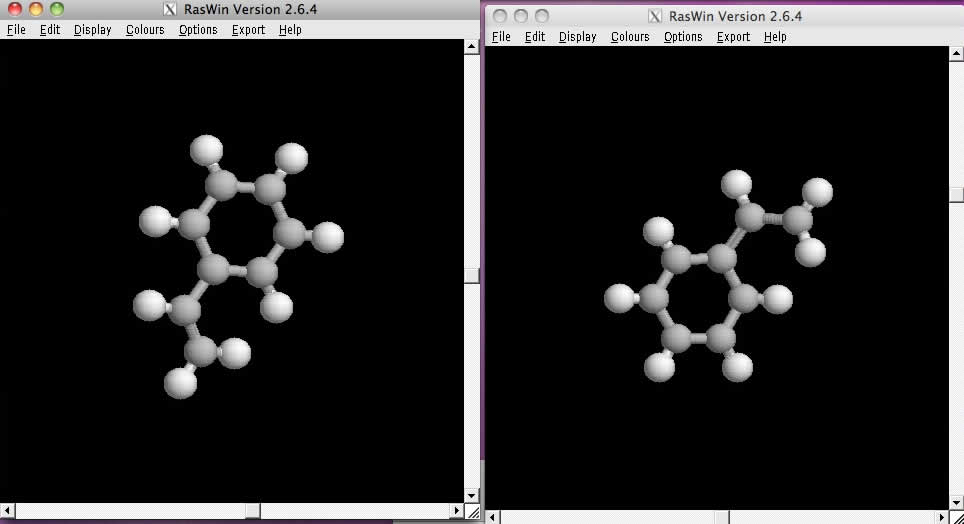
このように、Y-Molを使うと簡単に構造を作り出し、MOPACで半経験的分子軌道計算を行うことが出来る。
次にWinMOPACのFinal matrixのタブから、構造最適化が終わった構造をコピーする。
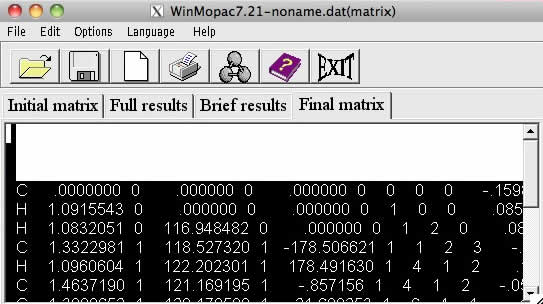
それをY-MOLのテキストエリアに(Bond inf.を残したまま)ペーストする。
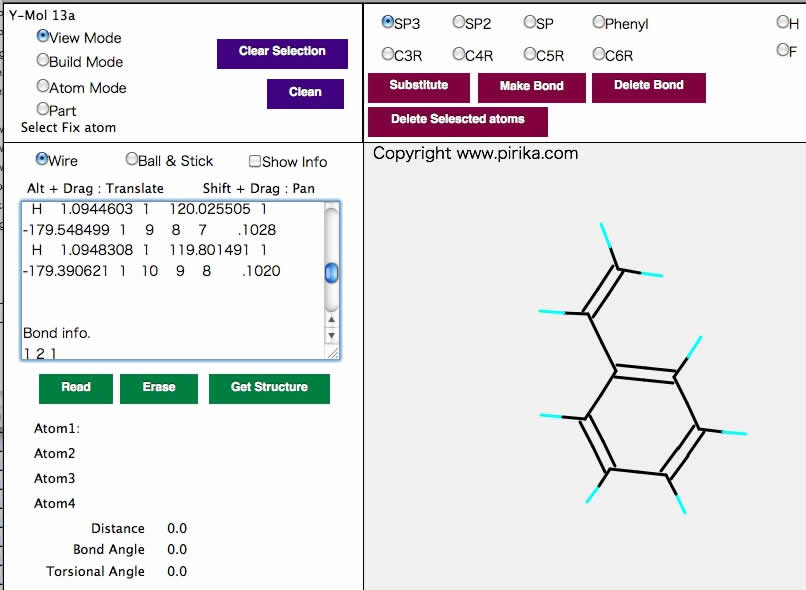
そしてReadボタンを押すと構造が最適化され終わった構造が読み込まれる。
そして、Part Modeを選択し、原子をクリックすると、原子の情報、結合長、結合角、ねじれ角などが計算される。
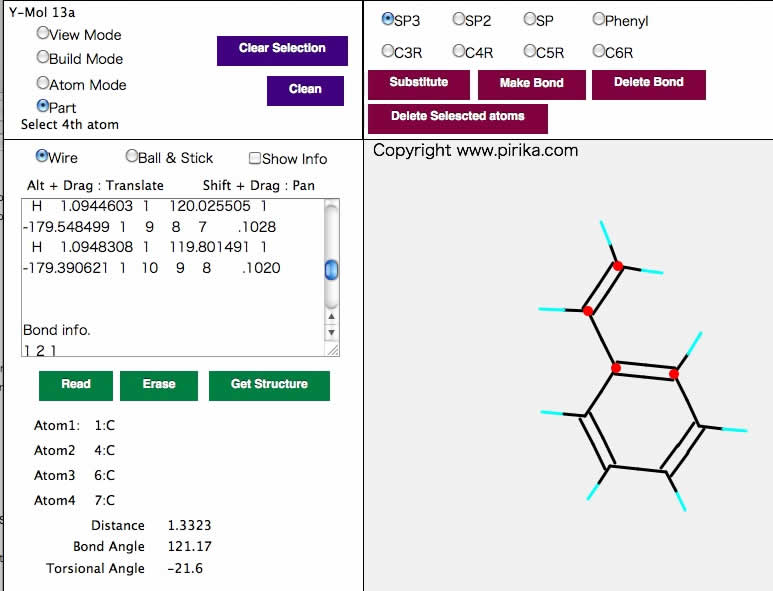
さらに、Atom Modeに切り替えて、変更する原子を全て選択してChangeボタンをクリックする。
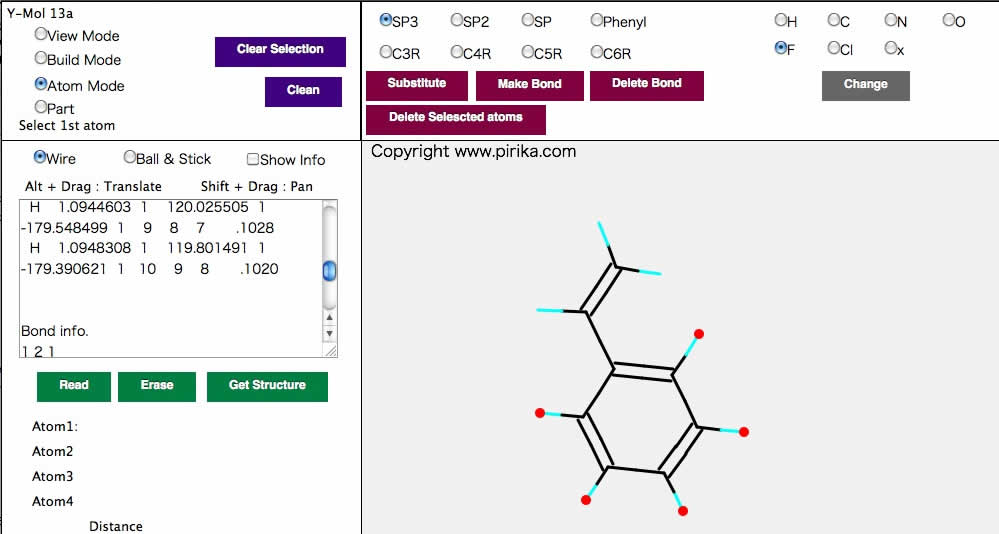
すると、水素がフッ素に置き換わった構造が得られる。その際には結合長は自動的に修正される。
これをGet structureボタンで取り出して計算などを繰り返すことになる。
リストに無い原子を使いたい場合には、xで置換する。xでマークされた原子はGet structureボタンを押すとXeと表示される。そこで手入力でXeを好きな原子に置き換え、Readボタンを押せば正しく表示される。(結合長の補正は行われない)
他のコマンドの使い方はYNUSの使い方を参照にして欲しい。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)