
2011.5.29
その他の化学トップページ> YNU-シミュレータ
HTNL+JavaScriptの手習で、昔作った(1990)Cのプログラムをブラウザー上で動くように改造した。現在では意味ないが、思い出の品として。
分子構造の最適化(分子力学計算)
自分が計算機化学を始めた頃(1987年頃)PCと言えばPC-9801、メモリーも640Kbype、ハードディスクも無く、1Mの3.5inchフロッピーディスクが始まりだった。IBMの大型計算機ではMOPAC3が動くようになったか、ならないかの時代だ。大型計算機に分子構造を入力する用のシステムが1000万と言われ当然そんなもの導入できない。ただ、その頃はQCPE(Quantum Chemistry Program Exchange)機構というところが、計算機化学用のプログラムのソースコードを配布していた。その時代の研究者はそうしたソースコードを読んで勉強したものだった。PCでもFortranは動かせたが、Fortranのフロッピーを入れると作業用のフロッピーは1枚しか使えず難儀したのを覚えている。PC-9801はN88-BasicをROMで持っていたのでBasicを使う限り、作業用のフロッピーは自由に使えた。そこで最初にやったのは、FortranのプログラムをBasicに書きなおすことだった。しかしインタープリター言語はどうしても遅いので、当時PC用に開発されたTurbo-CというC言語に移った。
その当時、MM2という分子力学のプログラムがあった。分子力学計算(MM計算:Molecular Mechanics)というのは、簡単に言えば、結合、結合角、ねじれ角をバネとみなして、標準値より小さければ、バネが反発し、大きければバネが縮む。そうした操作で構造を最適化する手法だ。計算は非常に速い(他の分子軌道法の構造最適化に比べて)し、計算できる原子数も多いので結構流行ったと思う。しかし得られるものが最適構造のみなのと、バネ定数が全ての組み合わせでもとまっているわけではないので、IRの振動(これはバネの振動に相当する)解析など一部の研究にしか使えなかった。自分は反応を扱っていたので、原子上の電荷だとか生成熱だとかを知りたく、分子軌道計算が必須だったので、PCでラフな構造を作成し、MM計算で構造を最適化して大型計算機でMOPACを計算するという作業をするPC側のプログラムを開発した。
そんな折、アメリカのカルフォニア工科大学(CALTECH)のGoddard教授がBioGraf/PolyGrafというMM計算を主体とするパッケージソフトを売りだした。Titanというスーパーグラフィクスワークステーションに搭載されたそのソフトはMM2で計算できない分子も計算でき、QEQという電荷平衡法で電荷も計算できるすばらしいソフトで計算機化学の一時代を築いたと思う。縁がありGoddard教授の所に留学する機会も得られ計算機化学に明け暮れした。そして、留学中にMac IISiを購入し、プログラム開発もMacに移行した。まー、そんな年寄りのノスタルジーは置いておいたとしても、最近はその道の専門家でもソースコードは読んだことが無い研究者がほとんどであろう。中身をブラックボックスとして受け入れて、使いこなしの方法を書いた書籍は沢山出版されている。ソースコードも持っていないから改良、改造もできない。この先どうなるのか他事ながら心配だ。
さて、当時書いたプログラム、BasicはC、C++、JAVA、JAVAScriptへと進化してYNUーシミュレータに搭載した。
繰り返しになるが、ほとんどの学生はWindowsのPCでアクセスしていると思う。Windowsの場合このシミュレータが動作するブラウザーはChrome、Safari、FireFox4.0だ。IE8以下はHTML5をサポートしていない。IE9はHTML5のサポートが完全でないので動作しない。Macの場合にはSafariを推奨する。iPadのMobile Safariでも動作する。対応するブラウザーを使っている場合にはリンクが下に表示される。リンクをクリックすると別画面が開く。こちらの画面で説明を読みながら操作してみて欲しい。
YNU-シミュレータを立ち上げたら、まず最初に最初の授業で伝えるパスコードを入力する。(パスコードがないと使える原子はH, C, N, Oだけになる。)
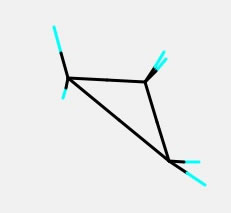
分子構造の作成のところで説明した、この構造を作ってみて欲しい。
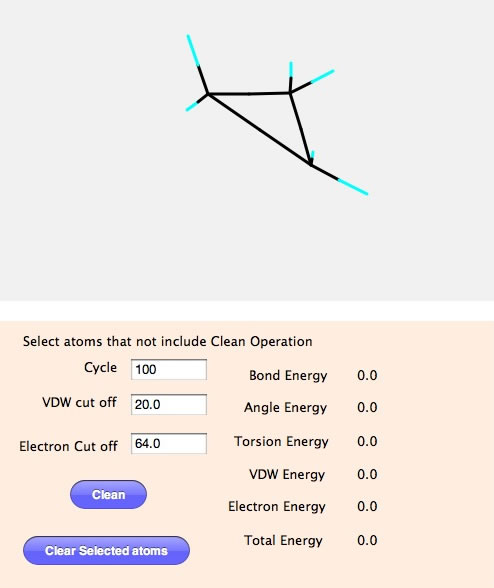
そしてMM計算のパネルに移る。
Cycleは何回計算を回すかを指定する。VDW cut off はファンデルワールスの力が及ぶ範囲を指定する。MM計算は結合、結合角、ねじれ角をバネとみなして、標準値より小さければ、バネが反発し、大きければバネが縮むとして計算を進めるが、このバージョンは結合の無い原子同士もファンデルワールスの力と電荷相互作用の力もバネとしてみなす。電荷も同様に力が及ぶ範囲を指定する。
そして、Cleanボタンを押すと計算が始まりすぐに最適化構造が得られる。
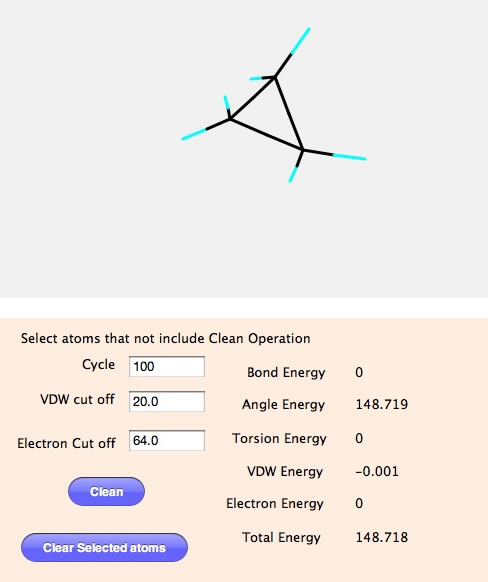
この場合は結合長とねじれ角は標準の位置に収まり、エネルギーは0になることが判る。結合角は3員環を作り歪が非常に大きいので148.7と大きな値になっている。手作業で構造の微調整を行うより圧倒的に簡単に構造が作れることが判るだろう。
このアルゴリズムは、まず結合のエネルギーを計算する。各原子をエネルギーが小さくなる方向の微小量を求める。順番に5つのエネルギーを計算し、5つの微小量の総和の方向に実際に原子を動かす。そこでこの場合、結合角のエネルギーを下げようと角度を広げる方向に一つを動かすと、他の角度はさらに狭まり、そちらからはさらに大きな角度を戻す力が働き、釣り合ったところで動かなくなる。そこで問題になるのは、5種類のエネルギーの絶対評価だ。角度を1度標準から変えるときのエネルギーと、結合長を0.02オンググストローム変えるときのエネルギーとの大小関係は?原子種が変わったときの評価は?と厳密なことを言うと、実は正しい構造とは言えない。ここで得られた構造を最新のMOPACやガウシアンで構造最適化したらもっと違う構造になるだろう。計算機化学を専門でやる人は信じないように。YNU-シミュレータの目的はあくまで、化学工学の学生が簡単に必要な計算ができて、実際の開発に役立つ計算可能量を得ることだ。
ここでの計算は電荷は入っていない。これについては電荷平衡法で説明する。
このMM計算では計算に含めない原子を設定することができる。どのような時それを使うか説明しよう。
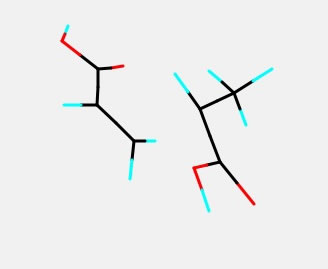
例えばアクリル酸の遷移状態(ラジカルが2重結合へ付加する反応)を計算し、上のような遷移状態が求まったとしよう。この構造を用いて分子を少し変更した構造を得たいとする。
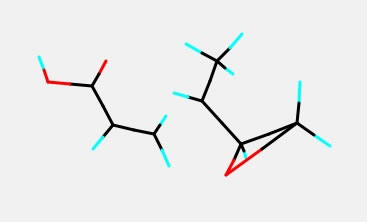
Builderを使えば簡単にカルボキシル基をエポキシ基に変えることができる。しかし、エポキシ環の構造は綺麗にしたいけど、遷移状態の構造は変えたくないといった場合がある。その時に最適化に含めない原子を下のように設定し、Cleanボタンを押すとそれ以外のところを最適化してくれる。
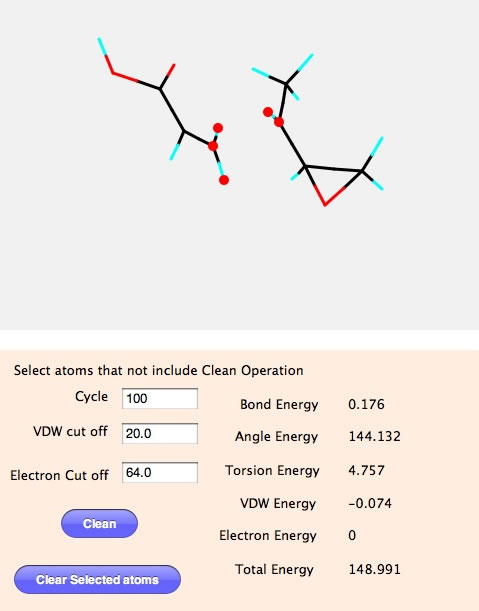
この構造を元にMOPACなどを用いて遷移状態を計算すると、非常に早く計算が収束する。
自分でこうしたプログラムを作成すると、非常に応用範囲も高い。例えば一つの分子の電荷まで含めて構造を最適化しておき、その分子を100個ランダムに3次元空間に撒き散らし、適当な方向に動かす。その時にVDWと電荷の相互作用だけを使った構造最適化を組み合わせれば、簡単な分子動力学計算が行える。実際には誘起される電荷もあるので、電荷が非常に早く計算できる必要がある。これについては分子の電荷計算で説明する。
昨日出来上がって、アップしたばかりなのに、今日にはGoogleなどでトップページにランクされている。いきなりすごいアクセス数だ。MM計算が廃れているという自分の認識は間違いらしい。
授業の最初にパスコードを与えるので、それを入力すればフルに使えるが、無くても機能制限はあるものの、動作する。プログラムはHTML5+CSS+JavaScriptを使って開発している。HTML5はまだ馴染みがないだろう。これはブラウザーのみでプログラムを走らせることのできる、新しい規格で今後のWebページを見る方法の主流になる。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)