
2011.5.29
その他の化学トップページ> YNU-シミュレータ
HTNL+JavaScriptの手習で、昔作った(1990)Cのプログラムをブラウザー上で動くように改造した。現在では意味ないが、思い出の品として。
分子の読み込みと表示
まず最初に必要な機能は分子の3次元構造を読み込む機能だ。
ここで扱うのは分子軌道法用の入出力ファイルなので、最も一般的なMOPACのファイルを読み込む機能をつけてみよう。(MOPACの説明は上記のWikiPediaの記事を参照のこと)MOPACの入力ファイルは、各原子をXYZの座標系で表した、カルテシアン座標系(直交座標系、Cartesian Coordinate、XYZ座標系、デカルト座標)と結合距離、結合角、ねじれ角で表した内部座標系(Inter Coordinate、Z-matrix)の2種類が扱える。詳しくはこちらの記事を参照いただきたい。
MOPACなどの分子軌道法パッケージソフトの開発の当初、計算機資源がまだ高価だったので、少しでも計算の量を減らす為、内部座標系(Inter Coordinate、Z-matrix)が使われたと聞いている。つまり、内部座標系で扱うと、最初の一つ目の原子は原点なので変数は無い。2つめの原子は原点からの距離だけで定義できるので変数は1つ。3つめの原子は距離と結合角なので変数が2つ。4つ目から変数が3つになる。 カルテシアン座標系では、1つ目から3つの変数になる。そこで変数が減る分だけ計算に有利になる。それに内部座標系では結合距離、結合角は原子のペア(と手が何本か?)が決まれば大体決まる。ねじれ角も1本目が決まれば残りの2本の結合はだいたい決まるので最適構造を探索するのも有利だったのであろう。ただし内部座標系にも問題点はある。アセチレンのように直線分子になると、結合角が180度になる。するとねじれ角が定義できなくなる。そのような場合には、ダミー原子を入れて結合角を減らしてやらないと計算エラーに成った。最近ではあまり気にすることなく、カルテシアン座標系を選んで良いと思う。(特に遷移状態の計算の場合はカルテシアン座標系の方が収束しやすいようである。)コンピュータの表示プログラムを書く際にはカルテシアン座標系の方が圧倒的に便利なので、読み込んだファイルが内部座標であった場合にはカルテシアン座標系へ変換しなくてはならない。意外とこれは面倒で、自分はMOPAC7というMOPACがまだオープンソースだった頃のFortranのプログラムを、C、JAVA、JAVAScriptへ書きなおして使っている。
それでは実際にやってみよう。
エタノールの内部座標系の座標は次のようになる。
C 0.00000000 +0 0.0000000 +0 0.0000000 +0
C 1.52065706 +1 0.0000000 +0 0.0000000 +0 1 0 0
O 1.43617510 +1 111.4497730 +1 0.0000000 +0 1 2 0
H 1.10730553 +1 112.3635704 +1 114.1418730 +1 1 2 3
H 1.10505636 +1 112.1389726 +1 -121.8276742 +1 1 2 3
H 1.09876132 +1 110.8669126 +1 -50.6926221 +1 2 1 3
H 1.09610606 +1 111.4898491 +1 -170.2492792 +1 2 1 3
H 1.09532834 +1 112.2350970 +1 69.1751491 +1 2 1 3
H 0.97504339 +1 110.7530221 +1 -54.0918564 +1 3 1 2最初の1つめは原子記号、次が結合距離。その後に0か1のフラッグが入る。これは1の時にその結合距離を最適化する計算を行うか、行わないかを示す。そのつぎが結合角、フラッグ、ねじれ角、フラッグと続く。その後に3つの数字があるが、これはテーブルの原子の番号を示している。2番目の炭素は1番目の原子と結合し、その距離は1.52065706であると判る。同じく3番目の原子は1番目の原子と結合し、距離は1.43617510で、3番目の原子、1番目の原子、2番目の原子で作る結合角は111.4497730であると定義される。これは定義上の問題であって実際に結合していなくても構わない。ただし、後に定義される原子は、既に定義されている原子だけを使って定義しなくてはならない。
MOPACのファイルを見ただけで、XYZ座標系かそうでないかを見分けるには、2つめの数字や3つめの数字が100以上のものがあるか、定義用の原子が3つ指定されているかで判断できる。この構造の部分をコピーしてメモ帳などにペーストしておく。
2012.6.16 同じソフトの読み込み部分を変更して、MOL(SDF)ファイルを読み込むようにしたプログラムはこちらから。
スタートアップ画面:
ここで授業の時に伝えるパスコードを入力する。正しければその後の計算は全て利用出来る。赤いPanel+ボタンを押す。(パスコードがないと使える原子はH, C, N, Oだけになる。)
ここで分子の読み込みのモードになる。(Get Structureボタンは正しいPassCodeが入力された場合のみ動作する。) ここに先程のエタノールの構造をペーストする。
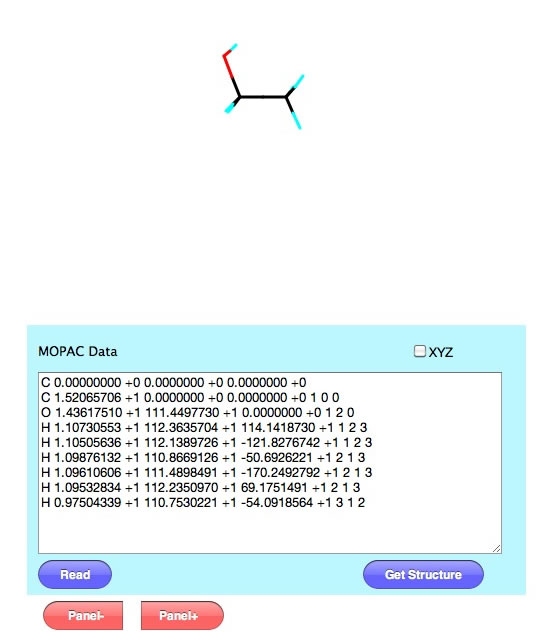
このデータは内部座標系なのでXYZにチェックは入れない。そしてReadボタンを押すと上に示すようにエタノールの分子が表示される。Panel−ボタンを押してもとのmain画面に戻る。
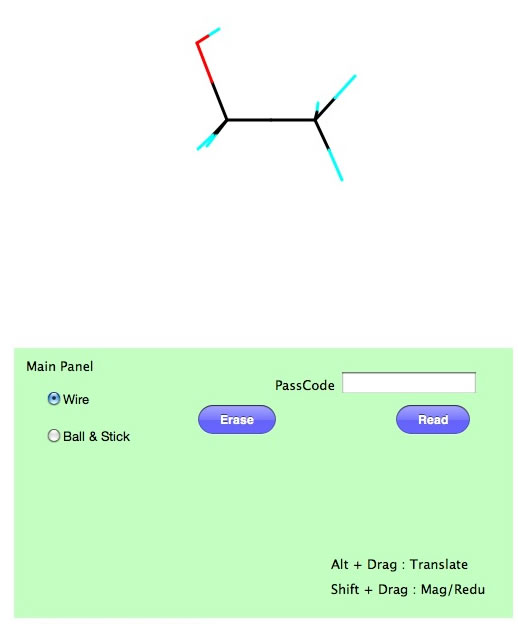
まず簡単な操作方法を説明しよう。分子の拡大/縮小はShift+Dragで行う。シフトキーを押したまま、分子のそばでマウスボタンを押したまま、マウスを左右に動かしてみて欲しい。移動量に応じて分子が拡大、縮小するのがわかるとおもう。iPadを使っている場合は、2本指をタッチして、片方を固定してもう片方を左右に動かしてみて欲しい。キーを何も押さずにマウスボタンを押したまま動かすと分子が回転する。また、Altキー(MacではOptionキー)を押したままドラッグすると分子が平行移動する。これはiPadでは3本指を使う。手で摘むような感じで動かす。
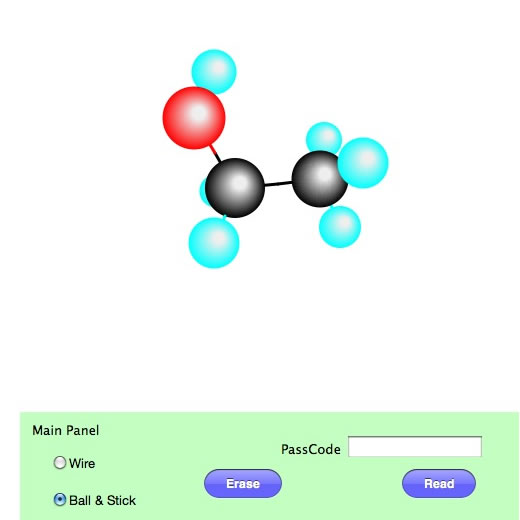
また、ラジオボタンでBall&Stickを選ぶと球-棒モデルが表示される。
これらの表示は3Dといいながら、実は完全な2Dグラフィックを使っている。擬似的に3D表示ぽく見えるようにするテクニックとしてパースペクティブ(消失点に向かって遠くのものほど小さく見える)と光があたったところから色を順繰りに変えるHTML5、キャンバスのコマンドを利用している。表示のテクニックは、まず、分子を読み込んで3次元座標を得たら、それは動かさない。そして回転と移動を定義する4次元の行列を用意する。分子を回転したり移動させたりする操作ごとにその行列を更新しておく。そしてその行列と分子の3次元構造をかけ合わせて、回転/移動させたときの3次元座標を得る。次にZ座標(画面に垂直方向)の遠いものから順番に描いていくと擬似的に3D表示ぽく見える。
次に、結合長、結合角、ねじれ角について見ておこう。
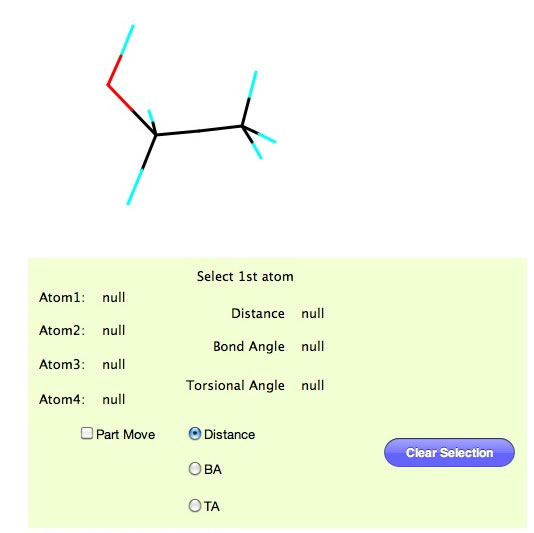
Panel+を何度か押して上図のパネルを選択して欲しい。
この画面で原子の上でマウスを押す。
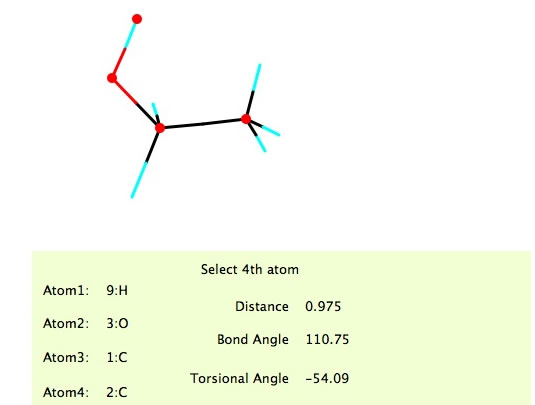
すると押された原子が赤くマークされる。原子が2つ指定されるとDistance(距離)が、3つ指定されるとBond Angle(結合角)が、4つ指定されるとTorsional Angle(ねじれ角)が表示される。同じ原子上でクリックされると選択は全て解除される。これらの計算は実際に結合していなくても行われる。通常のねじれ角は180を超える時には負の値で示されるので、ここでもそれを踏襲している。Part Move(部分の移動)については分子の構築のところで説明する。
一つ注意して欲しいのが、分子の読み込みは結合情報を使わない。そこで、3次元の座標を発生した後、標準結合長をもとに結合を推測する。この時に結合の次数(2重結合、3重結合)は考慮しない。しかし結合の次数が無いとその後の構造最適化の計算ではおかしなことになる。将来修正する予定ではあるが、自分で構造作成の所にあるように結合を作って欲しい。
以上、簡単に分子の読み込み、表示の部分を説明した。必要最低限の機能は実装していると思う。このように3次元空間に分子を表現出来れば、分子体積や分子表面積は比較的簡単に計算できる。分子がすっぽり入る直方体を考え、それに細かなメッシュを切る。ある点が分子中のどの原子からも結合距離以上離れていたらその点は、分子の外側だ。全ての微小点を分子の内側か外側かを決めてやれば、直方体の体積から、分子体積は計算できる。分子表面積については、Van der Waals のもの、溶媒が接触できる表面積(solvent accessible)とか、分子の表面をあるサイズの球を転がして測る表面積があるが、どのみち我々が扱う分子は絶対0度の真空中の分子ではない。暴れまくっている赤ん坊を体重計に乗せているようなものだ。あまり細かいことを言っても仕方がない。いわゆるVan der Waals の表面積を採用する。
分子表示を行う、他のソフトウエアー
分子を表示するソフトウエアーとしてネット上最もポピュラーなのはJMolであろう。様々な分子構造を読み込んで表示するJAVAのプログラムだ。自分のHPに組み込んで分子を見せることができる。FacioはOpenGLを使ったWindows用のソフトでGaussian、GAMESSなどの優れたフロントエンドとして利用可能だ。作者が日本人なのでマニュアル等を読むのも苦労が少ない。Chime (”チャイム”と発音する)はWebブラウザ上での分子の三次元表示を可能にするブラウザのプラグインとして開発されたが、最近はあまり見かけない。RasMolもほとんど更新されていない。
分子構造のフォーマット変換ソフトウエアー
YNUーシミュレータでは、入力のフォーマットはMOPAC形式に限っている。 そこで他のフォーマットをMOPAC形式に変換する必要があるかもしれない。その時にはhttp://ja.wikipedia.org/wiki/OpenBabelを使うのがいいだろう。
その他お金をかけられるなら、ChemDraw3D、ISISなどのソフトウエアーを使ってみると良いだろう。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)