
Many people say that what I am doing at Pirika is difficult and a hurdle to overcome.
However, it is important to go through high hurdles because it is impossible for an old man like me to force myself to jump over them.
For example, QSAR on the toxicity of dioxins (polychlorinated biphenyls) is difficult.
The Y-MB calculation for Estrone, the female hormone, and PCDD, the most toxic dioxin, is as follows.
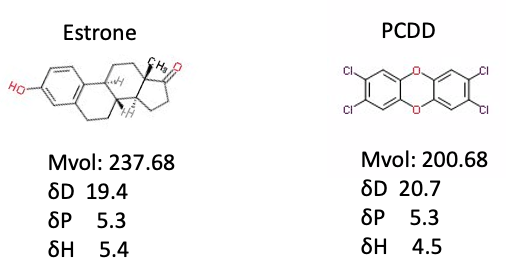
The shape of the molecule and the Hansen solubility parameter are very similar, so I think it may enter into the receptor that accepts female hormone and behave badly.
So far, anyone can do the calculation by purchasing HSPiP software.
However, as a developer of Y-MB, I am also faced with methodological limitations.
For example, if I want to count the functional groups, I need to break up the molecule. If I break the molecule into pieces, the information about where the functional groups were attached is lost. This is especially troublesome for aromatic compounds.
In the case of PCDD, both the left benzene ring and the right one are only recognized as tetrasubstituted benzene, so the calculated value will be the same even if the position of the chlorine changes.
The position of the chlorine changes the appearance and toxicity of Estrone. I need to improve the Y-MB.
It is a very high hurdle.
Currently, I am working on the construction and evaluation of Y-MB(2021) for ver.6.
Let’s try to apply this system to dioxin compounds.
The Y-MB estimates the physical properties of the compound if the SMILES structural formula of the compound is available.
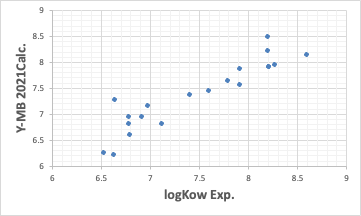
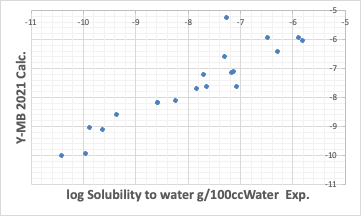
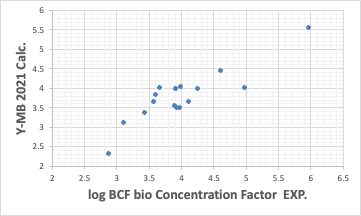
So far, the performance has been reasonably good. Such good estimation of thermodynamic properties is one of the features of Y-MB.
However, when the toxic equivalency factors (TEF) of dioxin are analyzed by QSAR using the Y-MB identifiers, some of the calculations do not match.
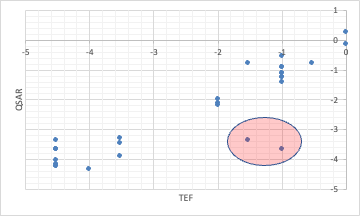
This is because PCBs with chlorine in the meta and para positions are called coplanar PCBs and are very toxic.
So, I’m looking for a way to go under the hurdle.
HSPiP has RDKit since version 5.1. It creates topological identifiers and 3D structures. So, I’m going to build a QSAR formula to estimate TEF by including the topological index and the result of CNDO/2 molecular orbital calculation from the 3D structure as well as Y-MB thermochemical properties.
In other words, if I give up the idea of doing everything on my own and go under the hurdles, my vision will expand.
If it doesn’t dissolve, it doesn’t matter if it’s poison. HSP
The shape of the molecule is also important. topological index Chi3n
The electronic information of molecules is also important. HOMO
Stability of the molecule. Heat of Formation
You can create a new QSAR model equation from these values
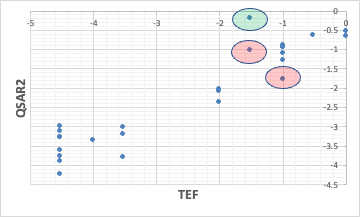
The coplanar PCBs that were off earlier have gotten better, but on the other hand, there is one more PCB that is way off.
It is important to consider why it is off, so I can allocate my time for developing my own software to it.
Usually, AI/MI/ML is done with only one of them.
However, if I could procure all the identifiers I need in one stop, I would be able to expand my horizon.
The following figure shows a CNDO/2 calculation of the 1,2,3,7,8-PeCDF with a large deviation. (This is not showing the computed results, but the computation is done on the browser, so it will take some time on slower machines.)
Rotate molecule: Drag with mouse (hold down mouse button); on iPad, hold down with one finger.
Move the position: Drag while holding down the Alt (Option on Mac) key. On iPad, move it with three fingers.
Zoom in/out: Hold down the Shift key while dragging; on the iPad, use two fingers to widen or narrow.