
2011.3.27
コンピュータの一番不得意なことは何でしょうか? それは幾何学じゃないかと私は思います。
コンピュータは身体と寿命の概念を持たないため、人間のように自分と比べて、大きい、遠い、長い、早いといった概念が絡んでくるものは不得意なのでしょう。
もともと数値しか扱えなく、目という器官がないので、化学の構造式をコンピュータで取り扱わせるのは結構苦労します。
自分も15年ぐらい前にWLN表記法を勉強しようとして挫折した記憶があります。
ただD体やL体、Cis-, Trans、E, Zなどはあまり関係ない化学工学の分野ではSmilesの表記法で十分なことも多いようです。
最近ではInChIやInChIKeyがコンピュータ用の構造式としては主流になりつつあるようです。
世間の動きはともかく、Pirikaでの分子構造の入力について紹介しておきましょう。
一番最初にプログラムを書いたときには、一番楽なのは原子団の数を自分で手入力してもらうやり方でしょう。
Pirikaの中のプログラムでも
Joback法を使った物性推算式
や
ポリマーの物性推算式
では未だにこれを使っています。
例えばWikiにあるJoback法の物性推算をプログラムにすると下のようになります。

試しにHTML5+CSS+JavaScriptのプログラムをつくってみたので試してみてください。
iPhone/iPod touchでは画面が小さいので別の設計にしました。
表にPOP-Upメニューを並べ官能基を選んでその数を入れるように変更します。この例では6種類の官能基が使えます。

官能基の種類を選んで、数を入れてCalc.ボタンを押します。
この形式だと原子団とその数をぺたぺたと貼りつけて、計算ボタンが押されたら、数式にしたがって計算を行い結果をテキストエリアに入れて返せば良いので非常に簡単にプログラムできます。
このやり方は分子が小さい場合にはいいのですが、ちょっと複雑になってくると正しく入れるのが難しくなります。
また表計算ソフトなどを使って管理するにはテーブルが非常に大きくなり不便であると言えます。
またプログラムの作者が意図しない分割法を使って誤った推算値になってしまうことがよくあります。
例えば、COOHというカルボン酸を、C=OケトンとOH水酸基と入力してしまうような失敗です。
そこでPirikaではごく初期から(1999年にPirikaを始めた頃)オフィシャルな入力フォーマットはSmilesの分子式を使いました。
Smilesの構造式についてはWikiの記事を参照してください。
このSmilesの式を原子団に分割することを考えます。
分割のアルゴリズムは自分で書いたものなので、意図しない分割になることはありません。
このやり方は分子に対して文字列のSmilesがひとつ対応するだけなので表計算ソフトで管理するには非常に都合が良いと言えます。
また原子団の取り方を変えた場合でも、簡単に追随できるので拡張性が非常に高くなります。
例えばアビエチン酸を例に説明しましょう。
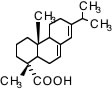
この中にどんな原子団が何個あるかを数え上げるのは大変で間違いも多くなってしまいます。
この化合物のSmilesの構造式は
C[C@](C(CCC(C(C)C)=C3)C3=CC1)(CCC2)C1[C@@]2(C)C(O)=O
になります。
データベース中にはこれだけが入っています。
これをテキストフィールドに入力してChemNeuroの分割法でConvertボタンを押すと下のようにどの官能基が何個あるか書きだしてくれます。
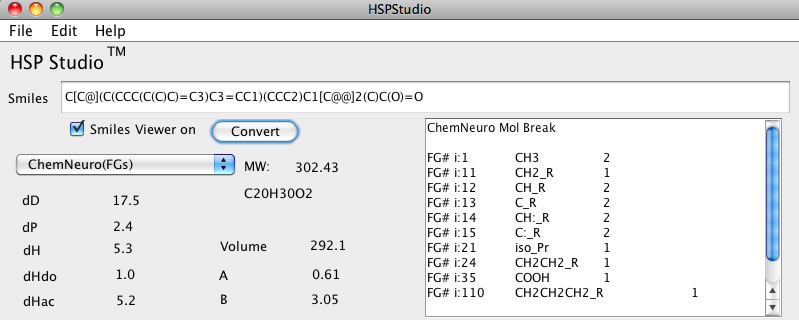
これを分割方法を変更して例えばJOBACK法を選択すれば、

Joback法の分割が適用されます。

慣れれば、Smilesの構造式を見ればどんな構造か頭の中で組み立てることができます。
そこで2−3年ぐらいこの入力方法が標準でした。
またPirikaでは分子を3次元座標でも取り扱います。
そこでMOPAC用のインプット・ファイルから原子団を数え上げるプログラムも開発しました。
それはさらに拡張されMOLファイルの読み書きにも対応されています。
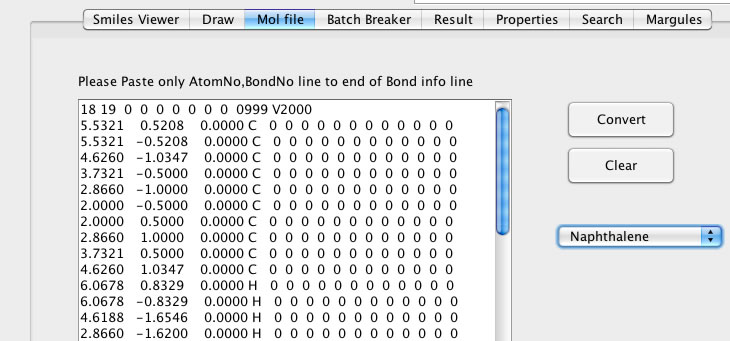
MOLファイルは薬学系ではよく使われているようですが、エクセルなどでは管理しにくかったです。正しい3次元構造は分子軌道計算をする上で大事なのだが。
現在では(2021.2)、MOPACのインプット・ファイル、Molファイルは、JSONで1行のテキストにして扱っています。コンバーターはpirika.comで公開していきます。
分子を描いてSmilesを得るプログラムを作ってみました。
Draw2Smiles:PC用
iDraw2Smiles:iPhone/iPod Touch 用
2004年ぐらいから、入力方法を分子を描く方法に切り替えました。
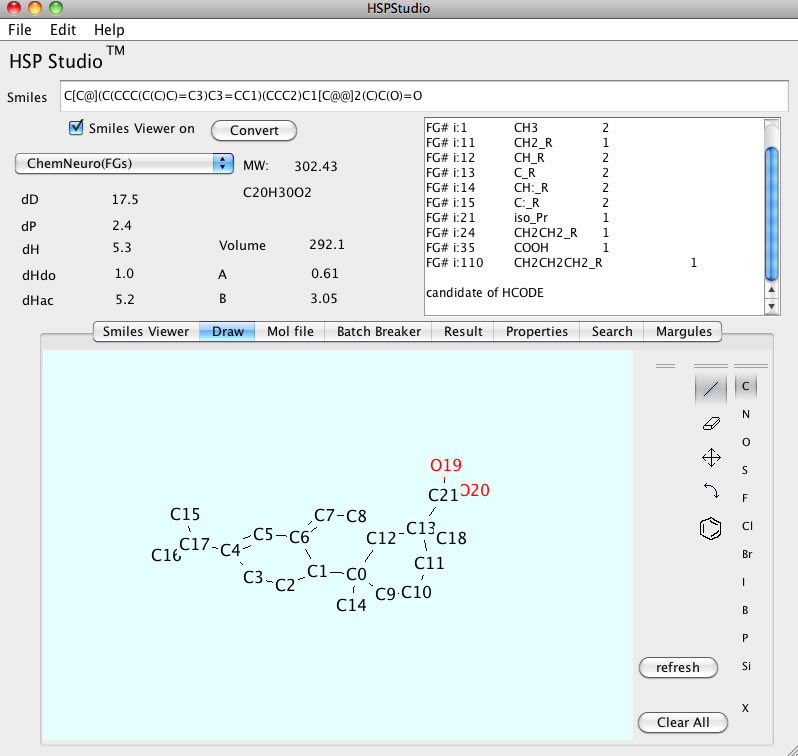
そのころから、Pirikaのアクセス数は飛躍的に増えているので、それなりに評価されているのでしょう。
この、分子を描いて、それから原子団を取り出す方法のメリットは、
意図しない分割方法は取れない。
使える原子の種類などの制限は描くことができるかどうかで決まる。
原子団だけでなく、原子団の結合情報まで利用可能。
などの点が挙げられます。
HTML5のプログラムはこちらから試してください。
JAVAのプログラムは現在は動きません
最終的には、Smiles、Draw、Molファイルが協調的に働くJAVAプログラムの形にしました。
HSPLightなどのデモを見てください。
大きな分子はSmilesから流しこみ、Drawで細かい修正します。Drawで行った修正はSmilesやMOLへ反映されます。
最初にこうした分子をコンピュータに認識させるプログラムを書いてしまえば、あとはすごく楽になります。
このようなことをしたいなら、まず原子団はなるべく広く認識出来るようにプログラムを書くと良いです。
広く認識したものを狭くするのは簡単ですが、逆は不可能だからです。
広く認識させようとすればするほど、例外が多くなりプログラムはどんどん複雑化します。
以前使っていたのと比べ現在のものは10倍くらいの分量になってしまいました。
原子団は3倍程度しか増えていないのに。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。