
2018.9.22
情報化学+教育 > MAGICIAN 養成講座 > 講義資料 > 第6b回 再構築学習法を取り入れたニューラルネットワーク法とドラッグ・デザイン
MAGICIAN(MAterials Genome/Informatics and Chemo-Informatics Associate Networks)
MAGICIANとは、材料ゲノム(Materials Genome)、材料情報学(Materials Informatics)、情報化学(Chemo-Informatics)を結びつけて(Associate)ネットワーク(Networks)を構築していかれる人財です。
概要
Materials Genomeを使って医薬品を設計することを考えてみましょう。
かと言って、医薬品のリード化合物を設計するのは実に難しいことです。
そこで、既にある医薬品の基本骨格を利用して、置換基を様々に変えた時に薬理活性がどう変化したか?を解析し、薬理活性を上げる置換基とその位置を特定する方法を学びましょう。
製薬会社は高度な計算機科学を駆使して行っている研究分野です。
これまでの研究蓄積と、高速なコンピュータを使って高精度な分子軌道計算などから“分子の遺伝子“を特定して、設計を行っています。
そのような方法は授業で教えられるものでも無いので、もっと簡単なMaterials Genomeを使ったやり方を解説しましょう。
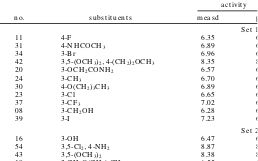
準備
まず、青山智夫先生の「ニューラルネットワークによる薬物の構造活性相関に関する研究」という学位論文をネットからダウンロードしましょう。
実は、筆者が使っているニューラルネットワーク法のプログラムは、青山先生が開発された再構築学習法を取り入れたものです。
共立出版の「階層型ニューラルネットワーク 非線形問題解析への応用」市川紘著(1993)にFortranのプログラム(PSDD: Perceptron Simulator for Drug Design)が記載されています。
昔はJCPE(日本化学プログラム交換機構:Japan Chemistry Program Exchange)からダウンロードできましたが、今は手に入らないようです。
化学系の学生が化学系のプログラムのソースコードを読む機会が失われたのは残念なことです。(ソースコードを読んで見たい学生はこっそり連絡をください。JCPEで配布されたコードは手元にあります。)
とあれ、25年以上前に開発されたニューラルネットワーク・シミュレータでDrug Designをどのように行っていたのか、青山先生の学位論文に記載の例題を実際に解析してみましょう。
ニューラルネットワーク・シミュレータ(NNS)自体は、最近、様々なものがネットからダウンロードできます。興味のある学生は、データセットだけ昔のものを使って、最新のシミュレータの実力を調べてみましょう。
例題解析
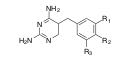
カルボキノン誘導体は、制ガン抗生物質マイトマイシンCに基づいて合成された化合物群で実際に医療に用いられています。
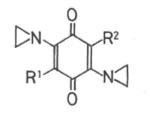
置換基、R1, R2を変えた時の生物活性値が求められています。
これらの化合物の活性値を予測するために、説明変数を準備します。(この部分が、こうした研究を行ったことがない初学の研究者やコンピュータしか知らないデータサイエンティストには難しいところになります。)
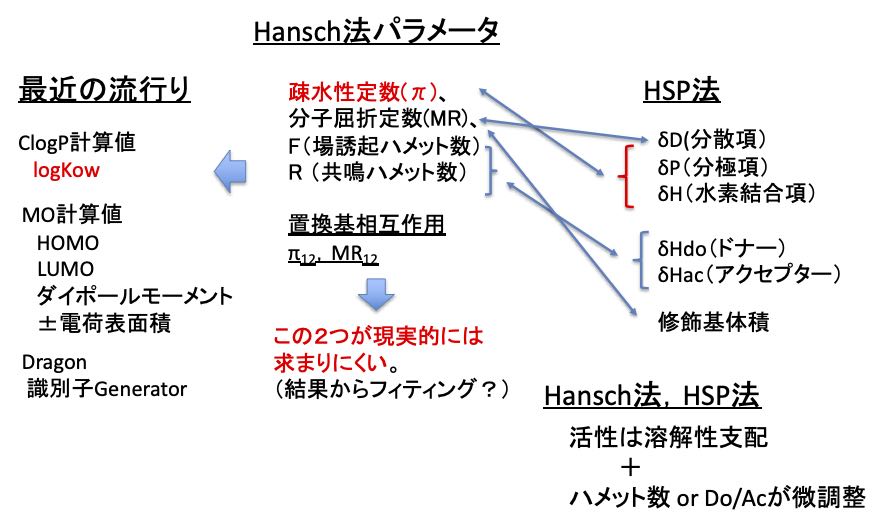
青山先生たちは、分子屈折定数(MR)、疎水性定数(π)、置換基定数(FおよびR)、立体効果と疎水定数を推定するためのMR1,2、π1.2の6種類の説明変数(識別子:Descriptor)を用いています。
これらの識別子の意味はおいておいて、まずは解析を行うためにデータを打ち込んでみましょう。
(近い将来、このような打ち込み作業はAIがやってくれるかもしれません。しかし現状ではまだまだ誤認識が多いです。汚いデータが入ってくると、修正する手間=クレンジングは、経験上、自分で打ち込む倍以上の労力がかかります。2024年、論文中のテーブル打ち込みはChatGPTがやってくれる時代になりました。ほんと助かります。)
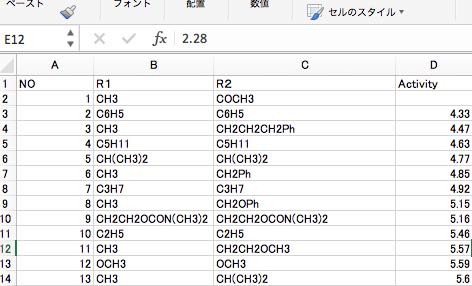
データ打ち込みが終わったら、まずは、重回帰計算を行ってみます。
ソフトウエアーは授業で配布したYSBを用いるか、Excelに搭載されている分析ツールを使ってもよいです。
Activity= -0.02169*MR1,2 -0.09473*π1,2 -0.4152480*π2 -0.274901*MR1 -1.771798*F -0.749842*R + 6.174982
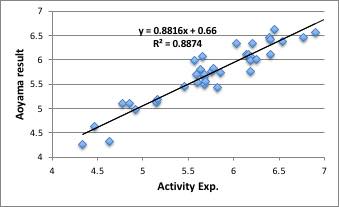
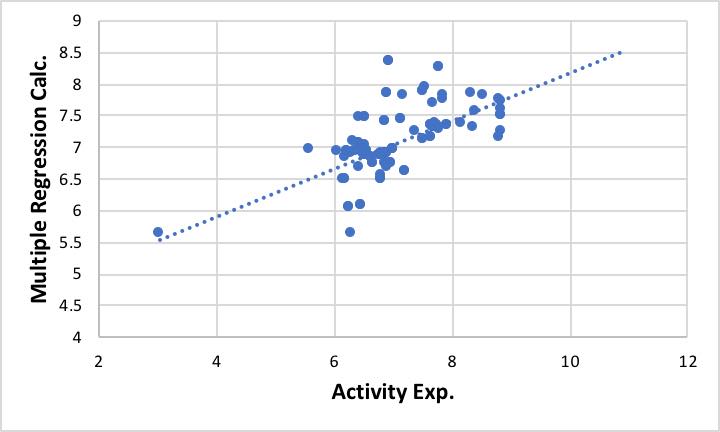
容易に重回帰式が求まり、それをグラフ表示すると下図のようになります。大きく外れるものは、打ち込み間違いのこともあるのでデータを見直してみてください。
何故、NN法で推算する前に必ず重回帰法で計算するのかというと、NN法は非常にフィティング性能が高いので、多少の打ち込みエラーがあってもフィティングしてしまい、どのデータが誤りかがわからなくなるからです。(例年いくら言っても端折る学生はいますが、痛い思いをして“学んで”もらうしかありません。)
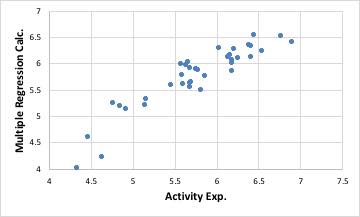
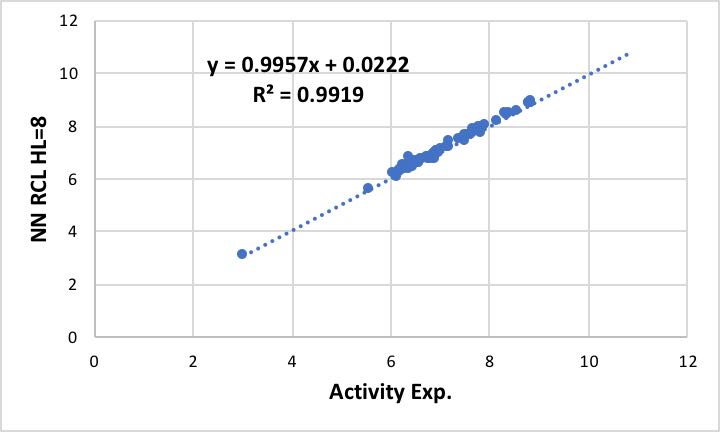
次に同じデータを用いて、ニューラルネットワーク法で学習させてみます。
学位論文では、入力層=7、中間層=12、出力層=1で計算していますが、私ののNNSは流石に20年以上改良を重ねているので、中間層は5でも十分高い精度で収束している事がわかります。
比較として学位論文中のデータをプロットすると下図のようになります。
通常はここまで出来上がると、Leave-n-out(L-n-o)解析を行います。
これは、学習データの中から、n個を外してモデル式を作成し、そのモデル式を用いて、外したn個を予測して予測性能を検証します。
しかし、私はL-n-oはあまり意味が無いように思っています。
例えば今回作成したモデルで、一番エラーの大きな5つを除いてモデルを作成し、その5つを予測してみれば、かえって誤差は大きくなります。
L-n-oが成立するのは、もっとビッグデータが存在して、その大多数が線に近いもので、少しの外れるものに使った場合だけではないかと思います。
それよりは、外れるものの特徴をよく考え、新たな識別子を導入するなどが実際には大事であると考えています。(例えば、フェニル基が入ると常に外れるなど。)
学位論文によれば、L-n-oの結果は良好だということで、ここまでできれば、次は置換基を片っ端から色々なものに変えて、活性が最大になる構造を探索するだけになります。
そこで問題なるのが、識別子の問題です。
青山先生たちは、分子屈折定数(MR)、疎水性定数(π)、置換基定数(FおよびR)、立体効果と疎水定数を推定するためのMR1,2、π1.2の6種類の識別子を用いています。
それでは、新しい置換基を考えたときに、その置換基の6つの識別子をどうやって手に入れたら良いのでしょうか?
分子屈折は屈折率、疎水定数はlog P(オクタノール/水分配比率)などから導き出される定数ですが、置換基定数はどうやって求めたら良いのでしょうか?
また、これらの定数は置換基が2つある時の定数で、どう取り扱ったら良いのかわかりません。
論文などを遡る必要があります。
この分野に明るければ、これらの定数はHansch法のパラメータであることがわかります。
「定量的構造活性相関 Hansch法の基礎と応用」C. Hansch / A. Leo 江崎俊之訳 地人書館
には、229種類の官能基の分子屈折定数(MR)、疎水性定数(π)、置換基定数(F、R)が記載されています。
それらがあっても、この学位論文の置換基を全ては網羅していません。
そこで新しい官能基を考えた場合、自分で6種類の識別子を作り出さなくてはなりません。
置換基のペアになった時の識別子の混合ルールを探さなくてはなりません。
そこで、最近の風潮としては、DFT使った高精度の分子軌道計算やって、HOMO, LUMO、ダイポールモーメント、分子の正負に荷電した表面積など計算結果を識別子にしてしまえ、という乱暴な方法に頼ることになります。
すると、スーパーワークステーションと高価なソフトウエアーが必要になり、一般人から遠い世界の研究開発になります。
それは、それで専門家に任せて、みんなが楽しめるマテリアルズ・インフォマティクス(MI)で医薬品を設計してみましょう。
MIの大元は、Chemo-Informatics(化学情報学)です。
化合物の情報は分子構造の中に存在します。
疎水性定数の意味合いは明瞭で、水酸基やアミド基など水溶性の高い官能基は値が小さくマイナスになります。疎水的な官能基はプラスになります。
分子屈折定数(MR1)は重原子の数が増えるにつれて大きくなる値なので、置換基の大きさ、重さを表しています。
F(場誘起効果)R(共鳴効果)はハメット定数に由来する定数です。
ハメット式は1935年ごろ、安息香酸類のイオン化に及ぼす置換基の効果として定数化されました。
昔のやり方は、ある置換基をm位やp位に導入した時の、安息香酸エステルの加水(加溶媒)分解速度から定数化するものでした。
当然ながら、複数の置換基が入り込んでくると、加成性はないのでぐちゃぐちゃになります。
近年は分子軌道計算などの結果と置き換わってきていますが、実際の設計段階では、置換基定数として与えられているF、R定数の方が逆設計上は便利です。
m位やp位の特異性は検討できませんが、溶解度パラメータを置換基の識別子として使う簡便な方法を紹介しておきましょう。
ハンセンの溶解度パラメータ(HSP)は、蒸発のエネルギーを、分散項(dD)、分極項(dP)、水素結合項(dH)に分割した3次元の溶解度パラメータとして知られています。
似たベクトルの溶媒は似たベクトルの溶質を溶解するので、医薬品の吸収などの解析に用いられています。
私も開発者の一員なので、様々な利用方法をHP(www.pirika.com)に記載しています。
この溶解度パラメータと分子体積を各原子団に対して決定します。
パラメータの決定には、授業で配布しているYMBを用います。
分子のお絵かきをして計算結果を取り出すだけなので非常に簡単に行えます。
使い方はこちらを参照。
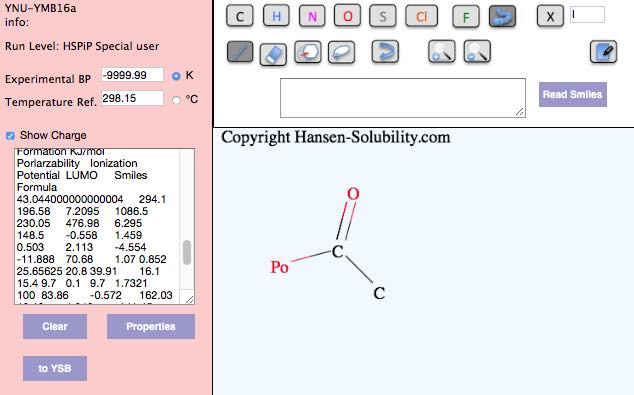
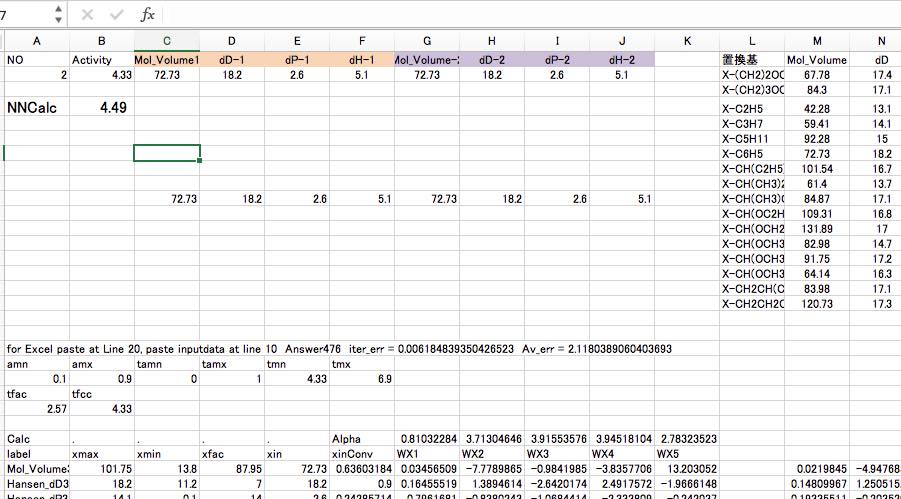
官能基にダミーアトム(下図でPoと表記)をつけた分子を描いてPropertiesボタンを押すとHSPも含め各種計算結果が出力されます。この結果をコピーし表計算ソフトにペーストしておきます。
このようにして、自分の用いたい置換基のHSPと置換基体積の一覧表を作成しておきます。
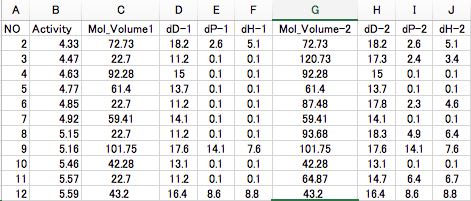
そして、先ほどの6つの識別子の代わりに、2組の [体積、dD, dP, dH]を識別子として使って上記のテーブルを作成します。
分散項(dD)は分子の屈折率と相関があるパラメータです。HSPの組みが疎水性を表現するので、誘起や共鳴は表現していませんが、医薬品の溶解性に関しては必要な情報は揃っています。
HSPiPというソフトを購入すれば、ドナー&アクセプターまで計算に含める事ができます。
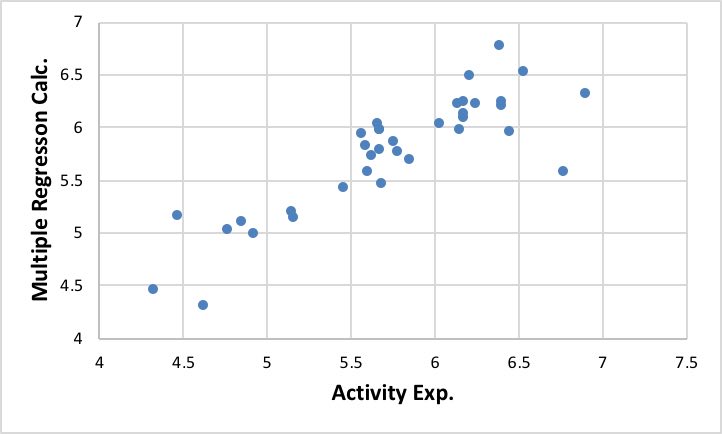
このテーブルを使って、まずはお決まりの重回帰計算を行います。
数点大きく外れるケースもありますが、大まかには悪く無い識別子であることがわかります。
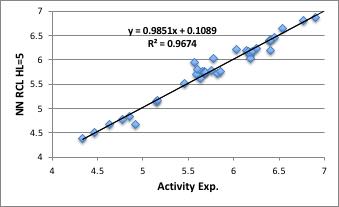
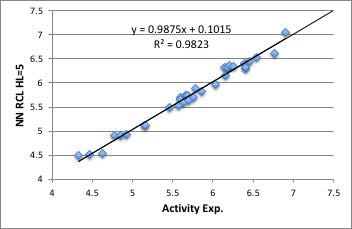
次に、このテーブルをニューラルネットワークに、再構築学習の頻度は1/500, 中間層=5で学習をさせます。
結果としては、下図に示すように良好に活性を表現できていることが分かります。
このHSPで収束したことは、次のことを示しています。
この化合物の活性は、溶解性が支配的で、置換基が電子を押したり、引いたり、共鳴したりはあまり大きくは関与していないと言えます。
この予測式が成立すると、置換基の識別子として[体積、dD, dP, dH]が得られれば、活性を予測でき、さらに活性の高いカルボキノン誘導体を分子設計できることが可能になります。
HSPは最近、dHをドナー性、アクセプター性に分割する試みもなされています。次世代バージョンではdHED, dHEAなどの指標も使えるようになります。
そうした新しい識別子も含めて置換基のデータベースを作成し、新たな医薬品設計を行うことが、普通のPCと授業で使っているソフトウエアーだけでできてしまいます。
それが、Materials Genomeの面白さであり、醍醐味です。
逆に言えば、高価なソフトと高価なハードウエアーが医薬品開発の参入障壁であった時代は終わったということです。
ちなみに筆者の作成したNNSは出力として、JAVA(C#)のプログラムを出力するようになっているので、プログラムが書ければ、置換基を順番に入れ替えて全ての組み合わせを計算して一番高い活性の構造を取り出すことも可能です。
MAGICIAN養成講座用には、Webアプリの雛形があるので、得られたプログラムをペーストすれば予測値はすぐに得られます。
また、Excelの計算式としても出力されます。
Excelのマクロが記載できるなら、置換基データを順に入れ替えながら、計算結果を取り出すことも可能です。
大事な点は、もし新しい実験を行って結果が出たら、すぐにニューラルネットワークを組み直せるかどうかです。
使えば使うほど、性能が高くなっていきます。
論文を書いたら終わりの大学の研究とは違い、企業の研究は競争に勝つために、開発スピードをどうあげていくか? が焦点になります。
そのために、Materials genomeやMaterials Informaticsを利用したいということを忘れてはなりません。
また、そうした再計算、再アプリケーション化が、その道の専門家に頼らなくてはできないのだとすると、スピードアップは期待できません。
自分でやるしか無いし、AIの教育係としては、そちらの方が適しています。
ともあれ、25年前にできていたDrug Designのシステムでも有効に働くことが再確認できたし、識別子としてもHSP(これも50年前のパラメータではあるが)が有効であることがわかりました。
それではもう少し複雑な系でも確認しておきましょう。
応用練習
Nonlinear Quantitative Structure-Activity Relationship for the Inhibition of Dihydrofolate Reductase by Pyrimidines
J. Med. Chem. 1996, 39, 3526-3532
この論文にPirimidine誘導体の薬理活性のデータが記載されているので、これを解析してみましょう。R1, R2, R3の置換基の種類と活性が記載されているので、置換基の[体積、dD, dP, dH]を全て計算しておきます。
(困ったことに、38. COH2CH2OCH3とか、62. OCH2Oとかいう多分誤植の置換基がありますが、これらは除いて考えます。)
この解析の場合は、カルボキノン誘導体の場合には無視できた、置換基の位置特異性(同じ置換基でも、m位、p位に付く時で活性が異なる)ケースが生じる可能性があります。
特にR1とR3は対称な関係の同じm位置換なので、3,4置換と5,4置換は学習させるデータに工夫が必要です。
まず、お決まりの重回帰計算を行うと下図のようになります。
一点大きく外れる化合物は、#10の3,5位に(m位に)CH2OHが付く化合物です。
#1の3,5位に(m位に)OHが付く化合物は今回検討されている化合物の中で最も活性が低いものです。
CH2OHとOHでは識別子の値が非常に似通ってしまうので、このような結果になったと思われます。
Activity=
0.00191*Vol-0.0367*dD+0.08908*dP-0.04885*dH+
0.00197*Vol-0.08230*dD+0.0089*dP+0.024390*dH+
0.00191*Vol-0.0367*dD+0.08908*dP-0.04885*dH+9.04893
重回帰式から明らかなように、3位と5位の計算値は同じになります。
例えばOHが付加する場合、[体積、dD, dP, dH]は、[16.5, 21.1, 21.9, 45.6] で同じですが、
3位、5位
0.00191*Vol-0.0367*dD+0.08908*dP-0.04885*dH=−1.02
4位
0.00197*Vol-0.08230*dD+0.0089*dP+0.024390*dH=-0.40
となります。
つまり同じ置換基であっても、付加する位置によって活性が異なる事が表現できています。
次にニューラルネットワークに、再構築学習の頻度1/500, 中間層=8で学習をさせます。
結果は上図に示したように綺麗に収束しました。
このケースでも、化合物の活性は溶解性が支配的で、置換基が電子を押したり、引いたり、共鳴したりはあまり活性に関係ない事が示唆されます。
これらの例のように、ニューラルネットワーク法を使ったQSAR(定量的構造活性相関)は2000年ごろにはブームでした。
しかし、ブームは急速に終焉し、サポートベクターマシン(SVM)、ベイズ統計などに移って行きました。
20年の時を経て、またディープラーニング法というニューラルネットワーク法がブームになっていますが、20年前は何故ブームが終わり、今回は一時期のブームで終わらないと言えるのか? 私の見解も示しておきましょう。
問題は、QSAR式の使い方にあると思います。
ある置換基の3つの組みが決まれば、活性が決まります。
置換基は、今回の場合3組の体積とHSP、[体積1、dD1, dP1, dH1], [体積2、dD2, dP2, dH2], [体積3、dD3, dP3, dH3]で表すことができます。
そこでニューラルネットワークの学習によって、活性と置換基の持つ識別子(体積とHSP)の関係を正しく表現できているのなら、予測性能も高いと期待してしまいます。
しかし多くの場合、この予測は裏切られます。前の6a回で述べたようにデータ数を増やせば、この過学習はある程度抑えることができます。
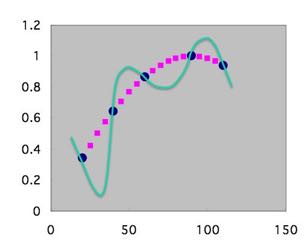
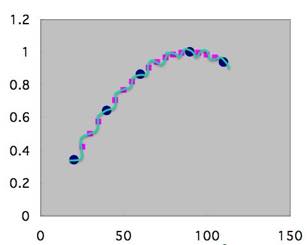
そこで、ビッグデータがあればニューラルネットワーク法のQSARでも予測性を高くする事は可能です。こうしたSinカーブであれば、自由にデータ数を増やす事は可能でしょう。
しかし、医薬品の開発では、置換基のHSPは同じで、体積だけ少し小さくというデータは取ることができません。ポリマーの設計の回で、量子化ベルトの話をしたが、飛び飛びの値しか取れないのです。
そこで、ある現象を、ある識別子で表現できた(収束した)後には、自己組織化ニューラルネットワーク(SOM)で検証します。
SOM: Self-Organizing Map法は、多次元ベクトルを2次元にマッピング方法で、1981年にコホネンによって開発されました。
このSOM法の解説は他に譲るとして、簡単に説明すると、似たようなベクトルは2次元上の似た位置にマッピングされると言う特徴を持っています。
つまり、今回3組の[体積、HSP]は12次元ベクトルで、この12次元ベクトルが、対称も含め104種類存在します。
この104種類の12次元ベクトルを、似たベクトルは似た位置にマッピングする事ができます。
口で説明すると難しいので、テーブルをコピーし、SOMのアプリケーションにペーストし、プログラムを走らせてみましょう。
104データを全て表示すると見にくくなるので、
活性が8.6以上のものをHH,
活性が7.74以上8.6未満のものをH
6以下のものをLL,
6.5以下6以上のものにLというラベルをつけた。
(以下の表示はPro版。Web版はより簡略の結果しか見ることはできない。)
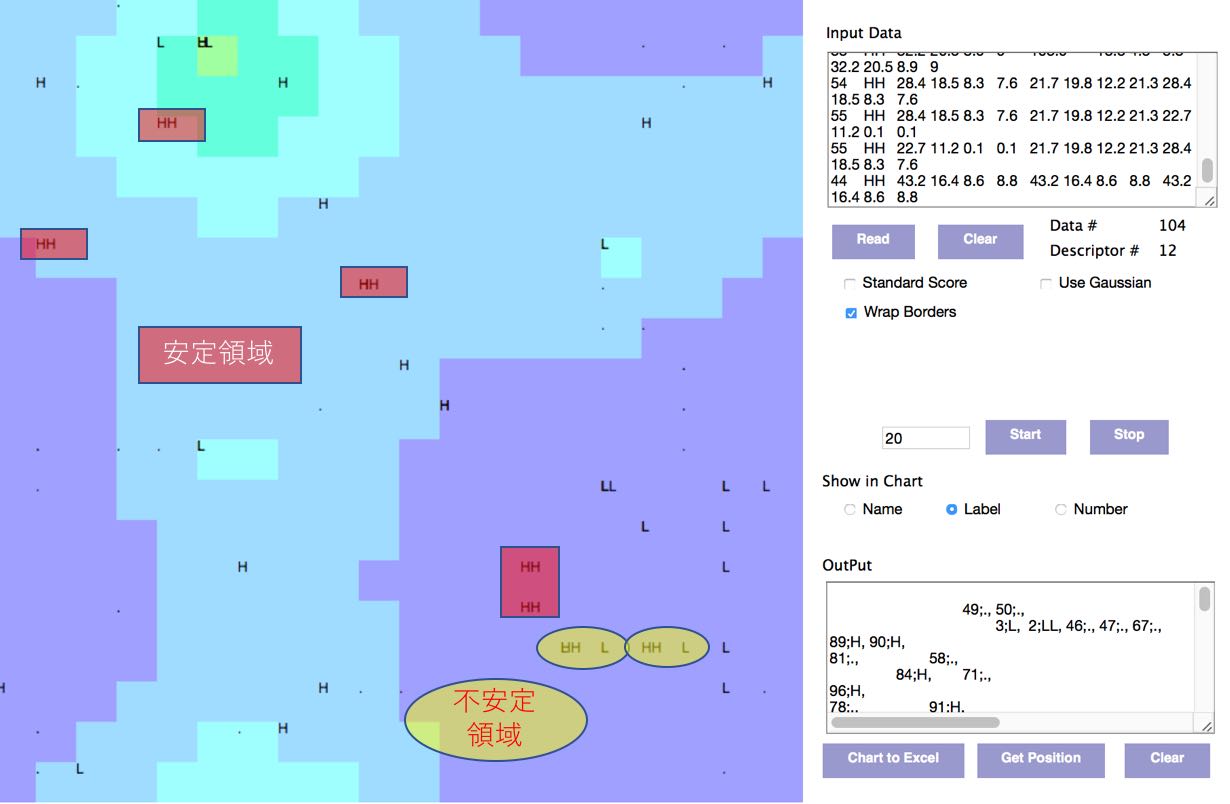
SOMの結果はキャンバスに表示されますが、Chart to Excelボタンを押し、結果をコピーし、エクセルにペーストする事ができます。
細かい検討はエクセルで行った方が便利です。
HHの近辺を調べてみましょう。
ほとんどのHHはその周辺に、LL, L, “.”、Hはいません。
ところが、2箇所ほどHHの周辺にLがくる領域があります。その近辺では活性はLからHHに急変します。
それを模式図として表すと下図のようになります。
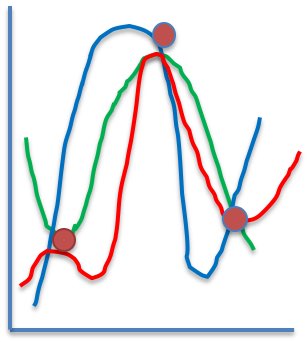
ニューラルネットワーク法で収束するという事は、活性が大きく変化する点を滑らかにつなぐ曲線をニューラルネットワークが見つけたということになります。
しかし、そのニューラルネットワークの中身はブラックボックスで、赤、青、緑のどのような曲線でつないだかはわかりません。
これらの点の中間ぐらいの修飾基があれば良いのですが、修飾基の「HSPを同じにしたまま体積を減らす」などという事はできません。
従って多くの場合は不安定領域では、たとえNN法がよく実験値を再現できていても予測性能は出ません。
安定領域にいる、HH、Hなどの修飾基を変化させた時の活性をQSARで予測して、開発する際の優先順位をつけて行くのが早道です。
つまり、定量的解析と定性的解析を組み合わせて評価していかないと欲しい答えは見つかりません。
ここで大事なのは、H、M、Lのラベリングは研究者の主観でつけたものだという事です。
つけ方次第で安定領域、不安定領域は変わります。
つまり研究者のセンスで考え方が変わる。勉強のしがいがある科学と言えます。
ディープラーニングなどの技術では、粗視化層の設計が重要になります。
画像解析の場合には、目、鼻、口とかで粗視化層を設計して行くようですが、構造式の場合はこうした自己組織化ニューラルネットワーク(SOM)で計算したマップを粗視化層に使うのも一つの考え方でしょう。
Indian Journal of Advances in Chemical Science 4(4) (2016) 374-385
なども、おもしろい論文なので、時間を見つけて書き足して行こうと思います。
雑記
ニューラルネットワーク・システム(NNS)自体は、今や非常に簡単に入手できます。
自分は、化学には化学特有の問題があり、それは数学者には理解できないと思っています。
そこで、青山先生が作られ、自分が成長させたNNSが、意味が無いとは思ってはいません。
もし、化学系の学生がシステム構築の方へ進むのであれば、最新のNNSに化学特有のオントロジーを組み込んで欲しいと思います。
NNSはそのうち良いものが使えるようになります。
プログラミングなどに興味のない大多数の化学系の学生は、NNSに食べさせる識別子をどれだけ合理的に作り出せるか? を自分の売りにして欲しいと思います。
3つの位置に100種類の官能基を順番につける。
全部で1000000種類のPirimidine誘導体を全部DFT計算して一番良いものを選ぶ。
30分で、100種類の官能基のHSP識別子を作り出し、1時間でニューラルネットワークに学習させ、3分で1000000種類のPirimidine誘導体を評価する。
DFT計算もできるに越したことは無いが、企業が欲しいのはどちらの人材だろうか? よく考えて欲しいと思います。
最近のAIブームで、化学系の学生も、計算機化学系を志望する事が多くなってきていると聞いています。
ハメット定数など、化学の深淵を覗かなくても簡単に識別子が手に入るのだから、勉強しなくても、楽して稼げる。
そんな、楽なやり方は、AIのもっとも得意とするところです。
近い将来、勝手にDFT計算しながら材料設計するAIができても自分は驚きません。
その時にAIに仕事を明け渡すか、自分の考えを組み込んだニューラルネットワークにAIアシストしてもらいながら仕事をするか。
人生観が問われているのでしょう。
ハンセンの溶解度パラメータ(HSP)を識別子に使うことの有効性を今回紹介しました。
HSPを原子団に割り振るのは、少しテクニックが必要だが慣れれば簡単です。
欧米の医薬品メーカーは全てHSPiPというHSPを扱うソフトウエアーは導入済みです。
最近、さらにHSPをMaterials Genome, Informaticsで扱いやすくした、CLI(Command Line Interface)バージョンのHSPの需要が高まっています。
このバージョンは、Pipeline Pilot を使って高速にMIを回すのに適しています。日本でもやっと引き合いが来始めたが、最初に購入するのはどこであろうか?
2020年から導入が急速に増えてきました。
CLIバージョンは個人で買える値段ではありませんが、学生バージョンのYMBが使いこなせれば、今回紹介したDrug Designぐらいは、より簡単にできるでしょう。
(逆にPipeline Pilot やMaterial Studioは個人で買えるようなソフトでは無いので、その中でどう使うのかは、自分は知りません。誰か、これらのソフトを寄付してくれないものでしょうか? とは言っても、自分はMacなので寄付されても動かないのですが。。。。)
情報化学+教育 > MAGICIAN 養成講座 > 講義資料 > 第6b回 再構築学習法を取り入れたニューラルネットワーク法とドラッグ・デザイン
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。