
2011.6.20
その他の化学トップページ >
MDシミュレーション
化学工学を理解する上で重要と思われる分子動力学(MD)のプログラムを試作してみた。分子動力学は簡単に言ってしまえば、分子はvという速度で飛び回っていて、1/2mv2というエネルギーを持っている。他の分子との相互作用はレナード-ジョーンズ・ポテンシャル(引力項の次数(q)を6乗、斥力項の次数(p)を12乗)という(6,12)ポテンシャルを使ってみる。これは分子構造の最適化で使ったものだ。そして温度が高ければ、早く動き、分子のそばに他の分子がいれば相互作用を受ける。計算の対象によっては電荷相互作用も大事になる。それについては電荷平衡法で計算する。速度論的な部分は、”分子のおもちゃ箱”というHPのJAVAアプレットを参考にした。
まず最初の例題は、箱の中に液体を入れて温度をかけてみよう。
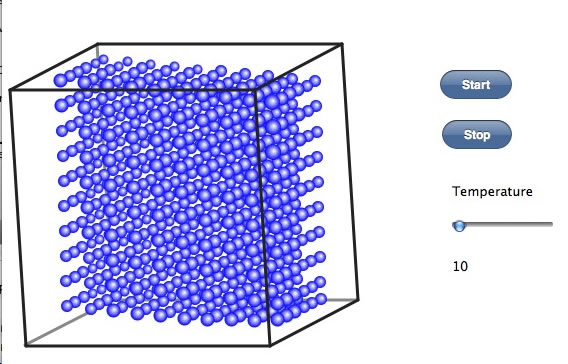
スタートボタンを押すと、箱の中に分子を600個発生させる。この分子は非極性分子で電荷は考慮しないとする。(温度の部分のスライダーはiPadやFireFox4.0ではまだサポートされていない。テキストフィールドが表示されるので値を入れてエンターキーを押す。)10℃では液体なので、すぐに落下して底のほうに塊になる。
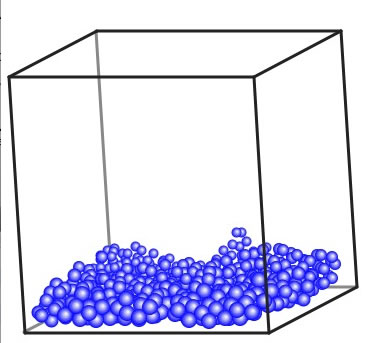
マウスでドラッグすると、箱を傾けた表示を見ることができる。液体と言っても動きが止まっているわけではなく、時々分子が飛び上がるのが見えるだろう。徐々に温度を上げてみよう。(いきなり温度を上げると、すごい勢いで飛び回る。これは、斥力が非常に大きくなり、核融合のような状態だ。)
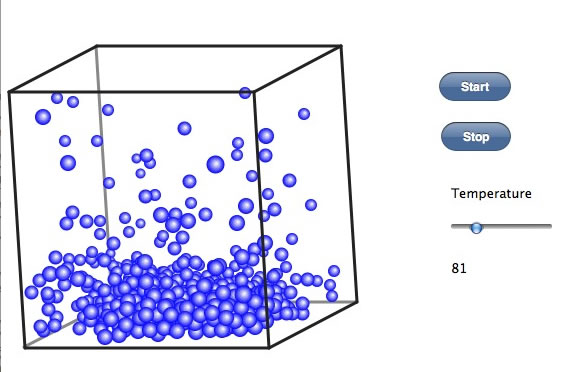
液体から飛び出した分子が、壁に衝突すると、化学工学上は圧力として観測ができる。時々分子同士が衝突して、速度を上げたり、並進したりする。これが熱伝導度して観測される。
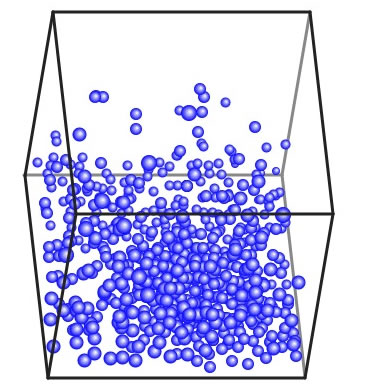
箱を回転させて上の方から見てみよう。液体は床に一様に広がっているのではなく、ある場所に盛り上がっているように見えるだろう。これは、この温度では動きが激しくなく、分子間力が高いので、凝集力が打ち勝っている状態だ。これは化学工学上は表面張力として観測される。
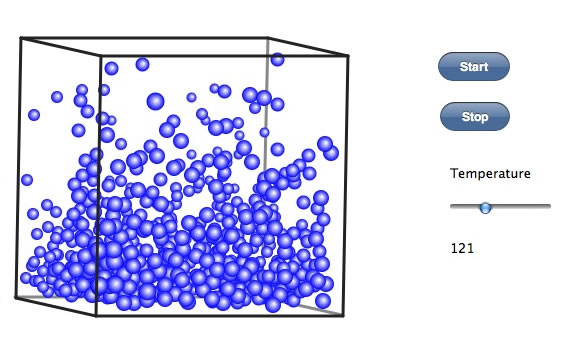
さらに温度を上げると、液体は床一面に広がり、分子同士の間隔も広がってくる。このことは、液体の密度は温度の上昇とともに小さくなることに対応する。
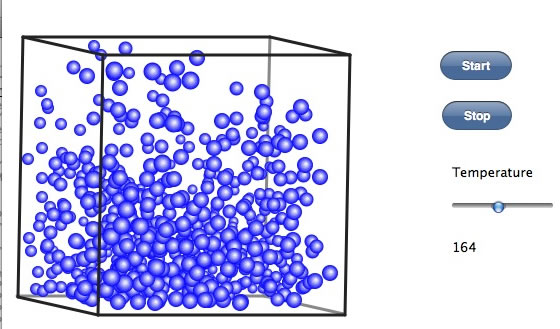
どこら辺が沸点であろうか? このシミュレータでは、気相中でも会合が多い。そこら辺のパラメータが悪いのか、ここが沸点と言えるような明確な点は現れていない。こうした会合が気相中、液相中の粘度として観測される。
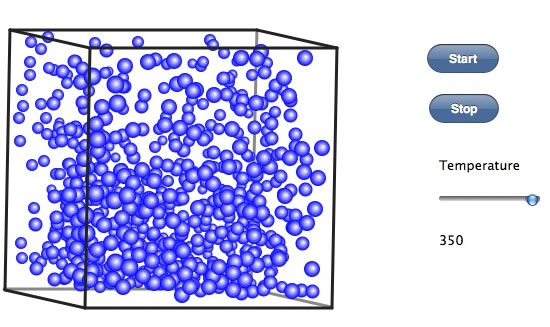
臨界点はこれらの分子が均等に広がったところで、あるので400℃ぐらいだろうか。
このように、MDを使うと、圧力、沸点、臨界点、粘度、表面張力、密度などの情報が得られることになる。多分MDの専門家からみると、まったくもっていい加減なソフトとお叱りを受けるだろう。しかし上に書いたような物性値とその温度依存性は、既に別口の物性推算法でかなり精度高く推算できてしまっている。どんなことが起きているのか視覚的に理解するソフトとしてお考えいただきたい。
次には蒸留を視覚的に見ていこう。
MDーシミュレータを立ち上げる。
対応するブラウザーを使っていれば上にリンクが表示される。
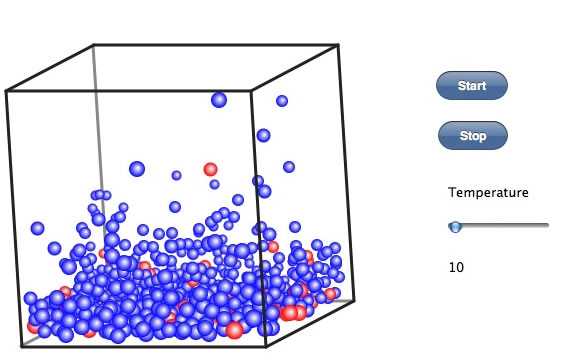
今度は、同じく非極性の分子だけど、分子量が約3倍の赤い球を10%ほど入れて同じことをやってみる。
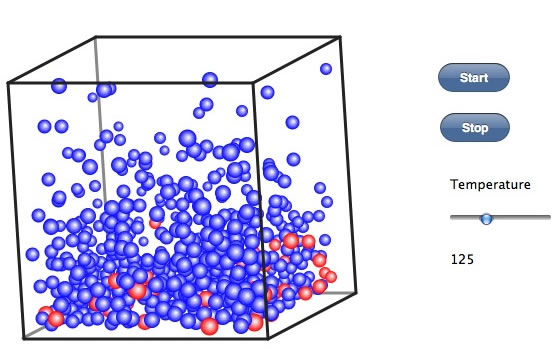
温度の上げ始めには赤い球も気相に出ている。そこでしばらく温度を一定に保つ。それが還流という操作だ。 その後、温度を上げていっても、赤いたまは中々気相にでない。この気相だけを集めるのが単蒸留だ。気相の上部に温度の低い部分を作って冷却してやると、分子の運動が減り、液体になる。こうした操作は、低い温度で行えば純度の高い青い球を集めることができるだろう。しかし効率は悪い。何度まで上げても気相部に赤い球こないか確認して、その温度をレポートに書いて欲しい。
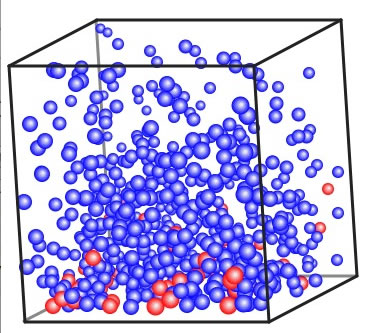
安定するまで少し時間を置かなくてはならない。
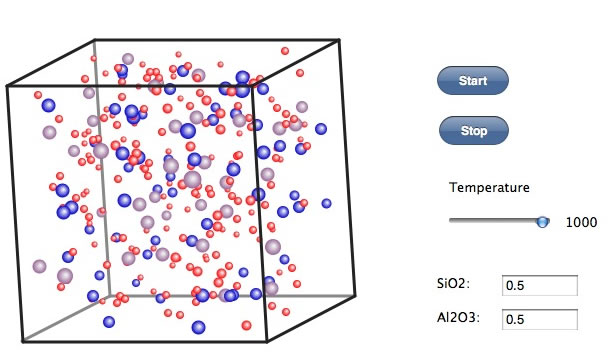
最近、新聞を賑わしているゼオライトを計算してみる。下のリンクを開きソフトを立ち上げてみよう。
この場合には電荷の計算が必要になる。電荷平衡法による誘起される電荷を計算するため、計算には時間がかかる。iPadでは無理だろう。まず温度を1000℃にすると、各原子は激しく運動して均一な分散状態になる。
700℃近辺にまで温度を下げると、Alと酸素が結合したような結晶核が現れるように見える。Siと酸素はまだ少ないように見えるが気のせいだろうか?
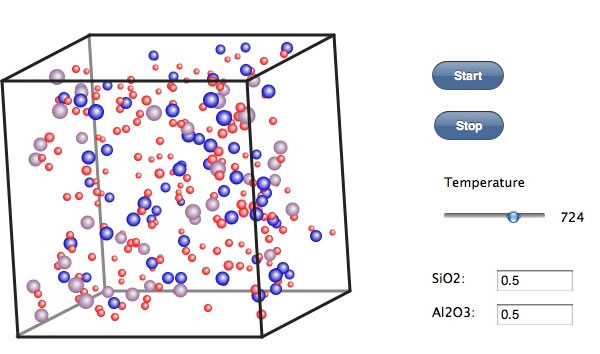
さらに550℃ぐらいまで下げてこの温度でしばらくキープする。徐々にクラスターが出来上がってくる。
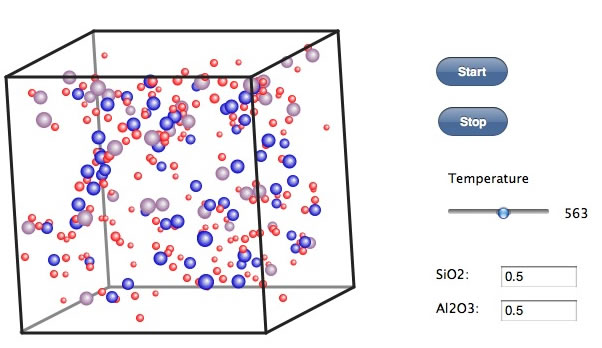
さらに400℃まで下げるとクラスターがはっきりしてくる。時々クラスターが融合して、また離れるのが観測される。まだフリーの酸素原子が多く存在する。Si, Al, 酸素の量比が合ったクラスターは安定だが、酸素が足りないクラスターや多すぎるクラスターは崩壊して新しいクラスターを作る。このくらいの温度を長い時間取るのが重要そうだ。
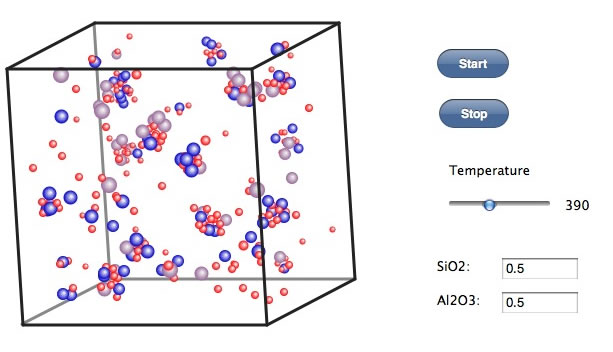
300℃ぐらいまで冷却するとかなり安定なクラスターになる。
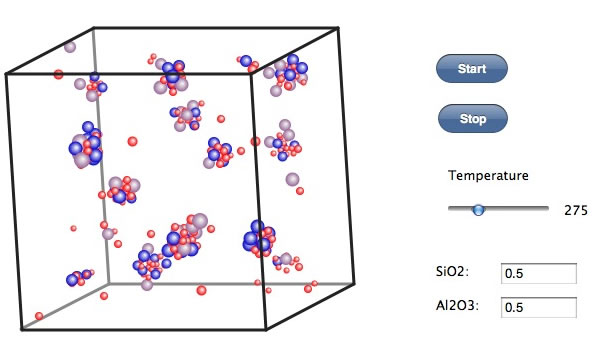
100℃くらいになると、ある程度規則性を持ったクラスターになる。
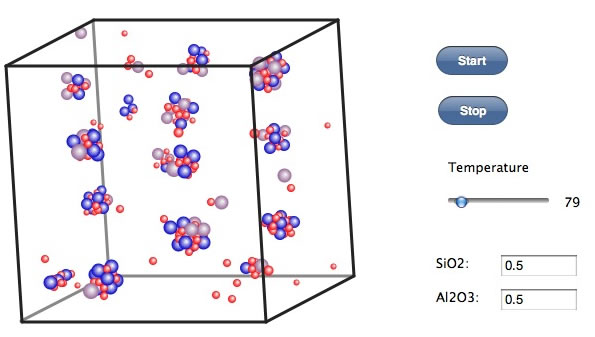
PCで計算するにはこのくらいの個数が限度であろう。(自分はiMac 27inch 3.06GHz Core 2 Duo で15分程度でこの結果になった。)しかし、こうしたクラスターが成長する際にはバインダーとして酸素が必要なこと。アルミが3価で真っ直ぐに伸びていかれないことからゼオライトは空孔を持った構造を与えるのだろう。温度の下げ方のパターンを変えたり、組成を変えるとできてくるクラスターの形も全然変わってくる。この空間にセシウムを閉じ込めるのだから、どんな組成、温度プロフィールでどんなモノが出来るのか予測することは非常に重要だ。試した結果のスクリーンショットと操作の概要をレポートして欲しい。
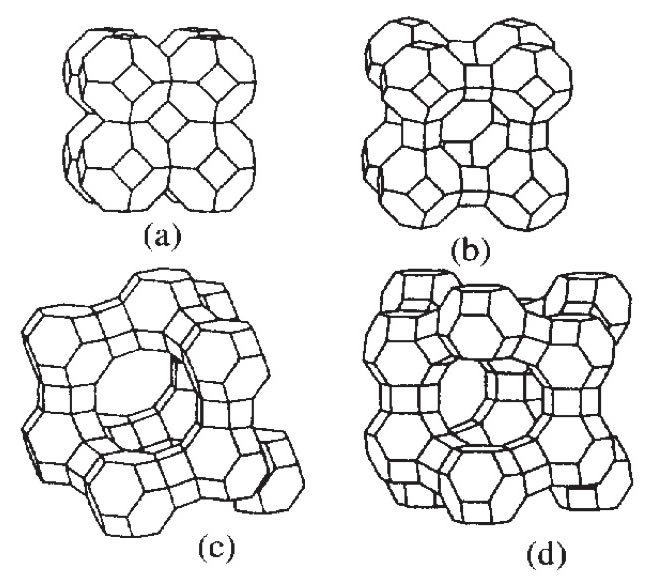
世界一の京速コンピュータを使って計算してみたいものだ。
何度も言うようだが、このソフトは分子動力学の専門家のソフトではない。温度と速度の関係も適当だし、電荷相互作用とファンデルワールスの力の関係も適当だ。特徴としては原子が移動した位置で、電荷平衡法で電荷を計算するので、計算は遅い。(だけど正確だと言えないところが辛いところ。) しかし、H(1)からLr(103)まで計算できてしまうのだから、それはそれで特徴かもしれない。
オリビン酸鉄(LiFePO5)を計算するとこうなる。下のリンクを開き、ソフトを立ち上げて試してみて欲しい。
Li原子は軽く、電荷も小さいので、動きやすい。ゼオライトよりも低い温度でキープした。最初に鉄(黒)とリン(緑)と酸素(赤)が構造を創りだして、温度を下げていくとLi(橙)がふんわりとくっついていくのが観察できる。一見してリン(P:緑)があまり均等に配置されていないようだ。自分の見た論文ではNbをドープすると結晶化が抑えられ、ガラス化するとある。実際に試しに計算して欲しい。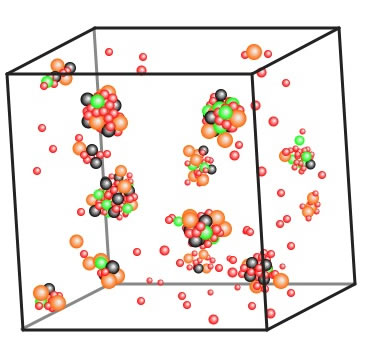
全原子対応版MDプログラム
こちらのプログラムは任意の原子を任意の割合で入れて計算できる。まず最初にパスコード(授業の最初の時に教える)を入力し、系の原子数を指定する。電荷計算を含める場合には500原子がいいところだろう。電荷計算をパスするなら1000原子まで計算できるようになっている。遷移金属などは価数が目的とするものでないかもしれない。色も大きい物は皆黒が入っている。おいおい修正しよう。ゼオライトにNaやKを入れたり、オリビン酸鉄にNbや他のものを入れて走らせて欲しい。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。