
2013.4.1
pirika.comで化学
>チャピエモン-3rd Pirika Origin (CPO)
> ハンセン溶解度パラメータ (HSP)
> 化学全般
> 情報化学 >物性化学 >高分子化学 >化学工学 >その他の化学 >昔のもの
>Pirika ツール群
ブログ
業務案内
お問い合わせ
>その他の化学 >MOPAC計算環境の構築
MOPAC計算環境の構築
最近、色々な大学の化学工学の授業の状況をお聞きする機会が増えました。
自分が結構驚いたのは、学生にガウシアンを使って計算させているところが結構あるという事です。
ガウシアンのようなAb-initio(非経験的分子軌道法)は、計算精度は高いですが、分子が大きくなると計算出来ない、コンピュータの計算能力が非常に高いものが必要になる、専門知識が必要になるなど、化学工学の学生には荷が重いと思っていたので、ものすごく意外でした。(他の大学の状況をお知らせ願えればと思います。)
一番大事な点は、おそらく卒業後、学生の多くは企業で研究を続けるのでしょうが、化学工学を専門とする企業の部署ではガウシアンが動作するマシン(ソフト自体も)が無いことでしょう。
化学工学の問題を解決するためにガウシアンのソフトとハードを購入して欲しいと費用対効果を上司に果たして説明できるでしょうか?
鶏が先か卵が先か?
そうなった時には、まずは簡単な半経験的分子軌道法で成果を出し、「ガウシアンを買ってくれたらもっと精度が高くなるんだよ」と説得するのが近道と思います。
その場合は自分でソフトを入手してインストールしなくてはなりません。
大学のように誰かがメンテしてくれて、自分は使うだけというのは許されません。
半経験的分子軌道法といっても、例えばChemOfficeなどに搭載されているプログラムを使おうとすれば、やはり50万近くかかってしまいます。
以前はMOPACという著名な半経験的分子軌道計算パッケージソフトがフリーウエアーで出回っていたのですが、Stewart先生が富士通で仕事をされている間に消え去ってしまいました。計算する分子を組み立てるソフトもフリーソフトは殆どなくなってしまいました。
そこで、現時点(2013.4.1)でフリーのMOPACを入手して、取り敢えず計算だけでも進められるような環境を構築する方法をまとめておきましょう。(企業のユーザーでお金の使える方はこの先は読む必要ありません。あくまでも貧乏人の為のMOPAC利用法です。)
半経験的分子軌道法MOPACの入手方法
大学の関係者(メールアドレス@ac.jp)は公式のMOPACを作られているStewart先生のHP(openmopac.net)から最新版MOPAC2012を入手できます。
大学を示すacがメールアドレスに入っていないと弾かれるので注意が必要です。
企業ユーザーは、日本では菱化(2021年違うかもしれません)から購入します。
インストールの方法などはダウンロードしたファイル群の中にマニュアルがあるし、ネット上にも情報はあふれているので、特に説明は要らないでしょう。
Mac版もWindows版もあります。
古いMOPAC7.1までは実行ファイル形式でStewart先生のHPからダウンロードできます。
学生の ac.jp で終わるメールアドレスを持っているなら使わない手は無いでしょう。
MOPAC606、pc-chem.info のSasakiさんが公開しているMOAPC6の改良版。これは完全にフリーなので企業ユーザーでも利用できる。ただし、MOPAC6をベースにしているので古い。
Macユーザーであれば、WebMo が公開しているMOPAC7がフリーで利用できる。
MOPAC7.21 ロシアの研究者がMOPAC7にビュアーをつけたものをWinMopacという名称で公開しています。
富士通が販売していたWinMopacとは別物なので注意が必要です。
これはXP, Vista, window7で動作しますし、macでもWineの環境が構築されていればwindowsが無くても動作します。
RasWinというビュワーも含まれるので手軽にMOPACでの計算を楽しみたい場合には最も適した方法かもしれません。MOPAC7といってもCOSMO法は含まれていないのでEPS計算はできません。
MOPACの走らせ方
MOPACの走らせ方は、どのバージョンを走らせるのかによっても異なります。GUIを持たずに直接走らせる方式の場合、ターミナル(Dos窓)などから
>MOPAC XXXX.mop
>MOPAC XXXX.dat
などと走らせる事になります。(>をDOSプロンプトと呼ぶ)この場合はMOPAC.exeがどこに置いてあるかをパスを切る必要があります。
パスの切り方についてはネットを探してください。
また、計算の対象の化合物のファイル名の拡張子は.mop .datが普通ですが、GUI付きのMOPACでは違う取り扱いのものもあります。入力ファイルをFOR005という名前で用意して
>MOPAC
などと走らせることもあります。
これはフォートラン時代の名残です。
ファイル名は常にFOR005でなくてはなリマせん。
このような場合には次のようにやると簡単です。
ファイルの修正の際に、.mopや.datという拡張子は嬉しくないので、拡張子は.txtにしてしまってバッチファイルで処理します。
(バッチ・ファイルについてはネットで調べてください)
@ECHO OFF
set a=Test
copy %a%.txt FOR005
MOPAC7
copy FOR012 %a%.arc
copy FOR006 %a%.out
del FOR005
del FOR012
del FOR006
del FOR011
del FOR016例えば上記のようなバッチファイルを書いて、mopac.batというファイル名でMOPAC7.exeと同じフォルダーにセーブします。
そしてTest.txtという入力ファイルを同じフォルダーに入れます。
そこでmopac.batをダブルクリックするとMOPACが計算され、Test.arcとTest.outが出力されます。
arcというのは計算結果のダイジェスト、outは出力の全部が格納されています。
繰り返し計算する場合には、最後の行の後に、
set a=Test2
のように続けて同じように書く。
GUIを持つものは、GUIソフトの使い方マニュアルをよく読んでください。
それでは、WinMopacで実際にやってみましょう。
(自分はMacなのでWine上で動かします。学生はWindowsにインストールしてやって見るように。)
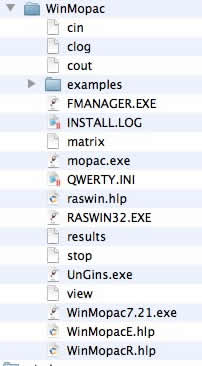
無事にインストールができると、ファイルが展開され、上記のようなファイル群が現れます。そこでWinMopac7.21.exeをダブルクリックします。
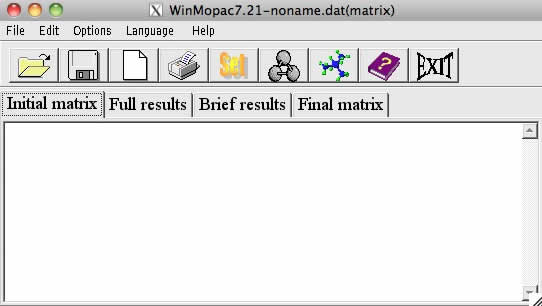
するとGUI画面が立ち上がります。
最初に例題を読み込んでみましょう。一番左のアイコンをクリックすると「ファイルを開く」モードになります。
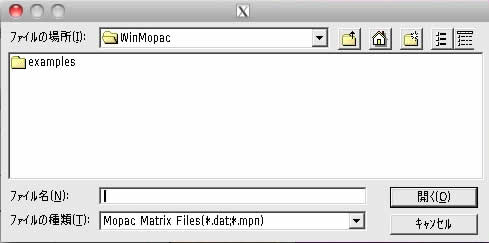
examplesのフォルダーの中に入り、
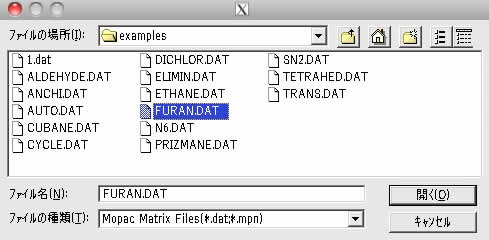
ファイルを選択して開くのボタンを押してみましょう。Initial matrixのタブの所にファイルが読み込まれます。
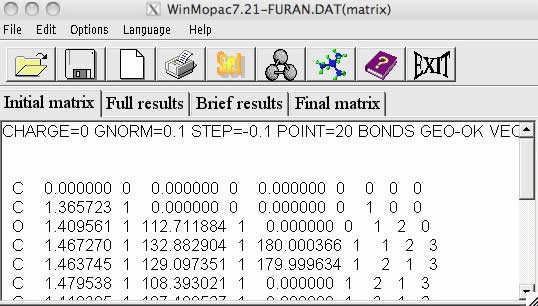
MOPACの入力構造は、最初1行目はキーワード行になります。
MOPACでどのようなキーワードが使えるかは、ネットで”MOPAC キーワード”と検索すればPDFで色々なバージョンのものが入手できるので調べてみましょう。
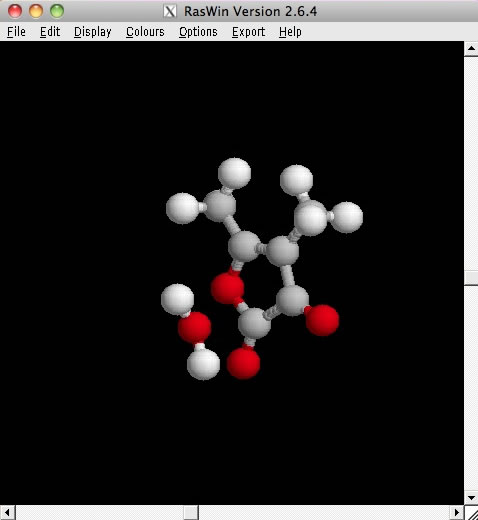
真ん中にある、原子が3つつながったアイコンをクリックすると、別画面に分子が表示される。これはRasWinという描画用のソフトで表示されています。
マウスを使って分子を回転させたりすることができます。
準備ができたら、RasWinアイコンの隣の分子の絵が書いてあるアイコンをクリックします。
上のようなダイアログが現れ計算が始まります。
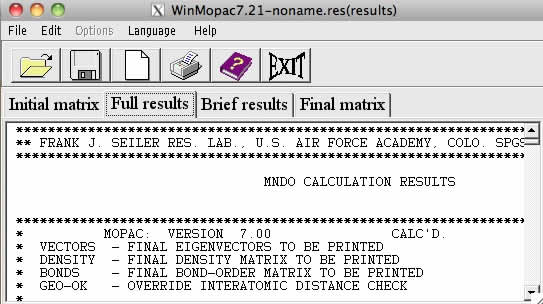
計算が終了すると、自動的にタブが切り替わり、Full resultsが表示されます。
これがMOPACの.outファイルに相当します。
通常のMOPACは.out, .arcを出力するが、タブにあるBrief resultsは.arcとは別物なので注意が必要です。
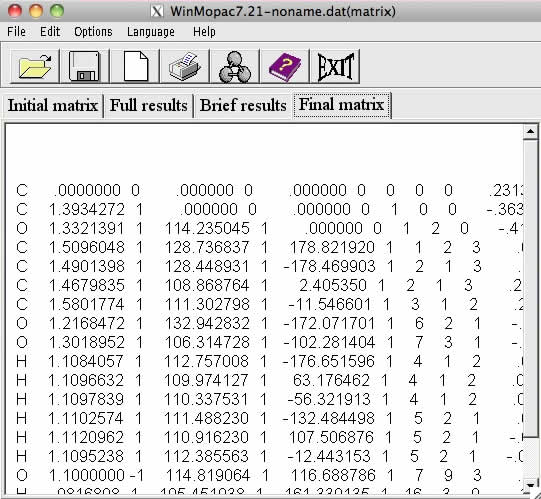
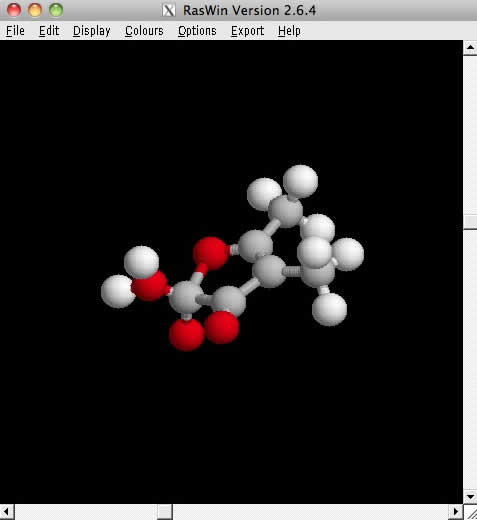
最終的に構造最適化が終わったものは、Final matrixに表示されます。再びRasWinで表示してみると構造最適化後の構造を見ることが出来ます。
後は必要であれば結果をフロッピーのアイコン(フロッピー・アイコンってもういいかげんにしない?)を押してセーブします。
このように簡単に分子軌道計算を行い、結果を見ることができるので使わない手は無いでしょう。自分で環境を構築し例題を走らせられるようにしてみましょう。
分子の構築
それでは、自分の計算したい分子はどうしたら準備できるでしょうか?
Chem3Dが使えるならそれを使って2次元構造から3次元構造を作りそのままChem3Dで分子軌道まで計算してしまいましょう。
ChemDraw3Dとほぼ同じように、分子の2次元構造を描いてMOPACの入力を得るプログラムを公開し始めました。
全くお金をかけられない環境なら、このPirikaのHPにあるYNUSというソフトを使って構造を作ってみましょう。
これはHTML5という技術を使って作ったブラウザー上で動作する分子の3次元構造エディターですが、お世辞にも使いやすいとは言いません。
さらに簡略版のY-Molというソフトも作ってみました。その使い方はこちらで説明します。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。