
2020.12.2改訂(2013.4.2)
その他の化学トップページ >
分子構築用ソフト、Y-Molの使い方
最初に左下のEnterボタンを押すと次のような画面になります。
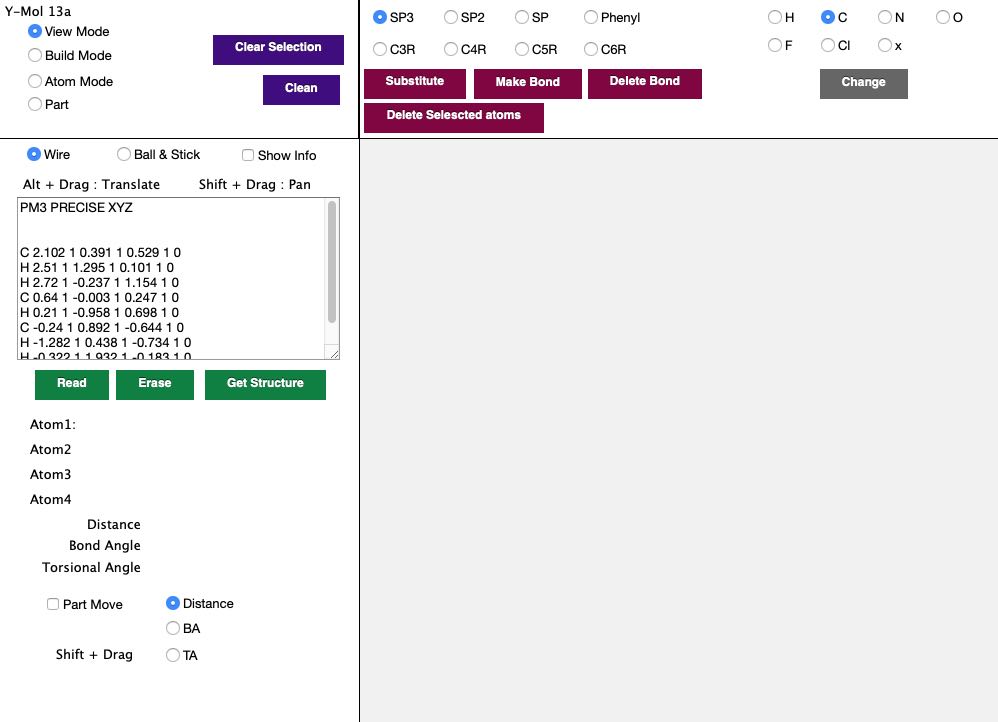
このY-Molは、HTML5+CSS+JavaScriptを使って開発しました。そこで、Javascriptというプログラムが動作するブラウザーを使えば、マシンを問わず動作させることができます。
頑張れば大きな分子を組み上げることもできますが、JSME+RDKitで分子構造を作る方が簡単です。
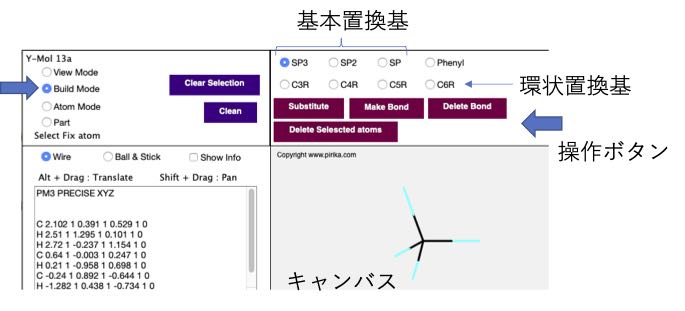
まず、左上にある組み立てモード(Build Mode)のラジオボタンを選択してください。そして、基本置換基でSP3が選択されていることを確認してから、薄い灰色の部分(キャンバス)をマウスでクリックしてください。
最初の1回目は炭素(黒)に水素(水色)がついたメタンが画面に現れます。
キャンバスでマウスをドラッグ(マウスボタンを押しながら動かす)すると分子が回転します。
Altキー(MacではOptionキー)を押しながらマウスを左右にドラッグすると分子が移動、Shiftキーを押しながらドラッグすると拡大、縮小されます。
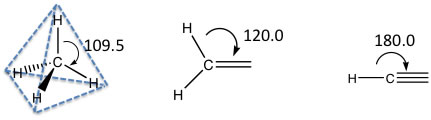
基本置換基のSP3, SP2, SPは次のような、ピラミッド(4面体)構造、平面構造、直線構造を示しています。
難しい言い方になりますが、原子は原子核とその周りを飛び回る電子からできています。その電子の飛び回り方が異なる、s軌道、p軌道、(d軌道、、)があります。
原子が他の原子と結合して分子になる時に、この電子の飛び回り方が変わります。例えばs軌道1つとp軌道3つから等価な4つの軌道(SP3混成軌道と言います)が作られるとピラミッド型の分子になります。s軌道1つとp軌道2つから等価な3つのSP2混成軌道が作られた場合には、平面型の分子になり、残った1つのp軌道が平面の上下に残ります。
簡単には、SP3は手が4本、SP2は手が3本、SPは手が2本と覚えておいてください。
本来は基本置換基だけでどんな分子でも構築できるのですが、環状化合物を作るのは大変なので、ベンゼン環と3-6員環の環状置換基が用意されています。
それでは、分子を伸ばして行きましょう。
表示モード(View Mode)以外のModeでは、キャンバス中の分子の原子をマウスでクリックすると赤く表示されます。操作の基本は元になる原子を先にクリックして、操作する原子を次にクリックすることです。
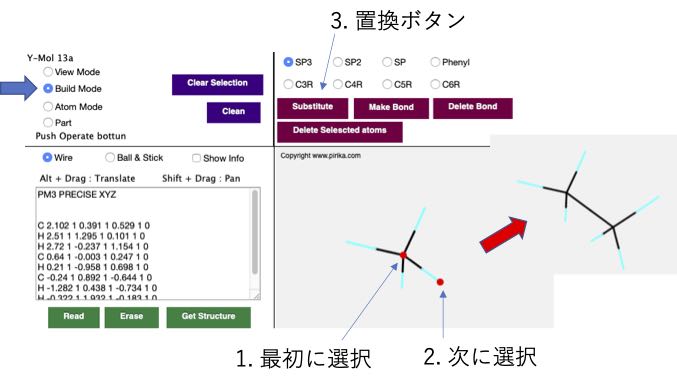
メタンからエタンの構造ができました。
同様の操作を繰り返すと鎖状の炭化水素化合物を作ることができます。
炭素が4つのブタンを作ってみます。
ブタンは真ん中の結合が自由回転できる結合になります。
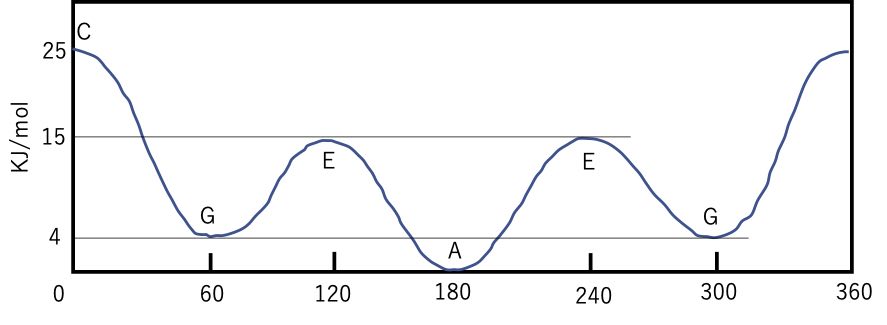
そこで両端の2つのメチル基がどういう向きかによって分子の内部エネルーが異なります。
0°から360°まで回転させると上のようなエネルギーダイアグラムが得られます。
C(Cis, シス)は真ん中の結合を画面に垂直に持ってくると、両端のメチル基同士が重なる配座の事を言います。下のデジタル分子模型はシス配座を取っています。ぐるりと分子を回転(マウスボタンを押しながら動かす)させどういう位置関係か確認してみてください。針金モデルでは両端のメチル基は遠いように見えるかもしれませんが、実際の立体障害は25KJ/mol程度と大きなものになります。
シスから60°回転したG(Gauche, ゴーシュ)の配座は、右に回しても、左に回しても同じものになるので2つ存在します。シスの立体障害は緩和される方向なのでエネルギーは下がります。
シスから120°回転したE(Eclipse、エクリプス)の配座は、メチル基-水素の立体障害が2つあるので、また少しエネルギーが高くなります。
そして、シスから180°回転したA(Anti、アンチ)はエネルギーが一番低くなります。
Drag=回転, Drag+Shift キー=拡大、縮小, Drag+コマンドキーかAltキー=移動。
次は、このデジタル分子模型で、こうした配座異性体の構造を作る方法です。
形は気にしないで、炭素を7つに増やして、nーヘプタンを作ってください。
ここまで、できたら3次元構造を取り出す(Get Structure)ボタンを押します。
この3次元構造データはMOPACという半経験的分子軌道計算ソフトの入力データになっています。
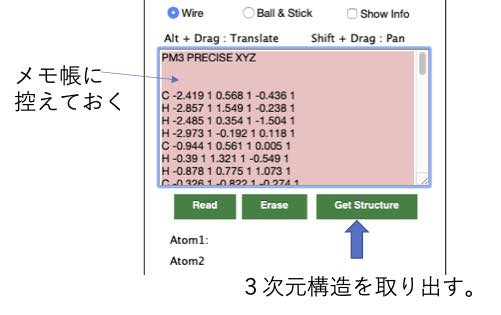
取り敢えず、テキストエリアの中身を選択し、コピーし、メモ帳などのテキスト・エディターに貼り付け、セーブしておきましょう。 何あったときに構造を戻せます。
さて、nーヘプタンは6個の炭素-炭素結合を持ちます。
この結合は単結合で、環に属していないので自由に回転させることができます。
実際に回転させて見ましょう。

まず、部分操作(Part)を選択します。
そしてキャンバス上の原子を4つクリックします。
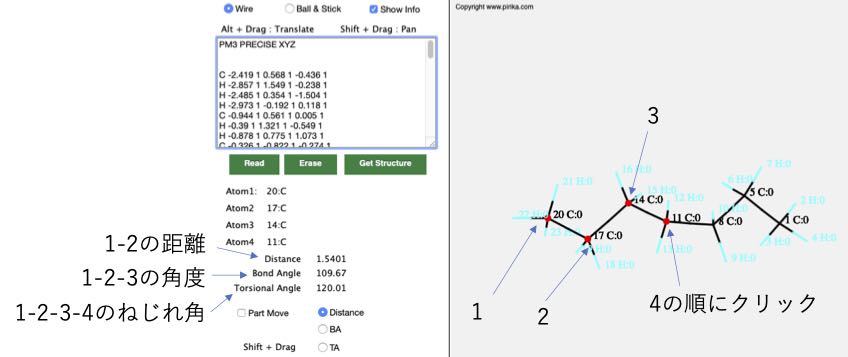
結合の回転は、2番目に指定した原子と3番目に指定した原子の間で行われます。結合を回したいだけなら原子を2つ選択するだけで良いではないかと思うかもしれません。
キャンバス中でマウスをドラッグ(マウスボタンを押したまま動かす)してみてください。
そして2番目と3番目に選択した原子を画面上で重ねあわせてください。
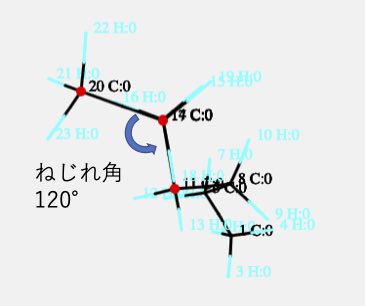
最初の3つの原子が作る平面と、後の3つの原子が作る平面の2面角(ねじれ角)が120°であることがわかります。ただ感覚的に回すのでは無く、シス、ゴーシュ、エクリプス、アンチの配座を意識して結合を回転させるためには原子を4つ指定する必要があるという事です。
それではこのねじれ角を動かしてみましょう。部分を動かす(Part Move)にチェックを入れ、ねじれ角(TA)を選択します。そして、シフトキーを押しながらマウスをドラッグしてください。
3番目から先に付属する(動かされる)原子が皆赤くマークされます。
ねじれ角が120°から180°になるまで動かしてみましょう。

この操作を各炭素-炭素結合に対して繰り返します。
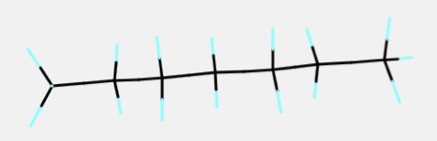
ねじれ角を全て180°にすれば全てアンチ配座になるので一番安定な構造になります。
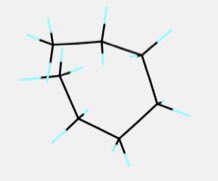
分子の末端同士がぶつかるような構造は不安定な構造になります。
それでは、次に、2,2,3-トリメチルブタンの構造を作ってみましょう。
一旦消去(Erase)して初めから作成しても良いですし、n-ヘプタンの構造を一部流用して作成しても良いです。
2,2,3-トリメチルブタンは次のような構造になります。
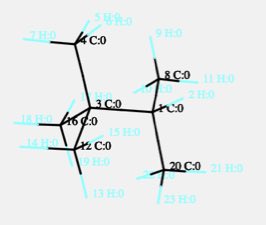
炭素-炭素のねじれ角はいくつあるでしょうか?
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。