
2013.4.10
pirika.comで化学 > 化学全般 > 高分子化学 >
ラジカル重合の開始反応
ラジカルの生成速度
有機過酸化物はラジカルの発生源としてラジカル重合には無くてはならない原料だ。しかし事故も非常に多い。
「レゾルシン製造工場で爆発事故」2012年講義資料より
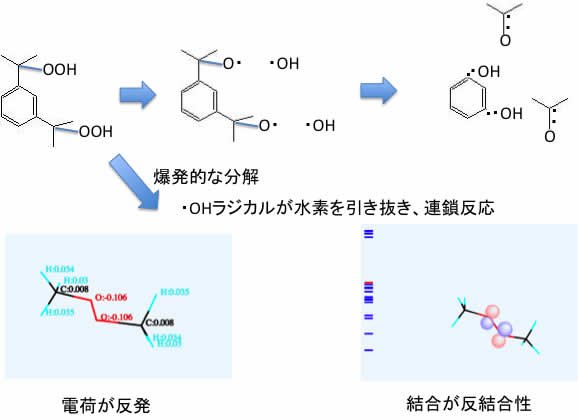
ChromeなどHTML5準拠のブラウザーを使っているなら、上にmethyl peroxideのCNDO/2の計算機結果が表示されるだろう。
表示されない場合
これは計算された結果を見ているのでは無い。
CNDO/2の計算と表示プログラム、t-butyl peroxideの構造データがPirikaサーバーから送られてきて、使っているコンピュータ上で実際に計算したものを見ている。(iPad, iPhoneでは少し計算に時間がかかるかもしれない)
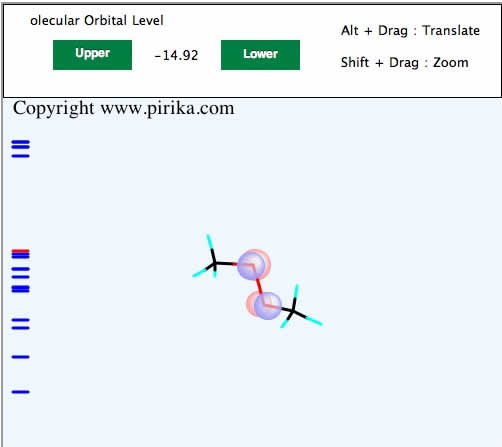
左の部分に青い線があるだろう。これが分子軌道のエネルギー準位というものだ。
最初に見た時にはそのエネルギー順位はHOMO(Highest Occupied Molecular Orbital)を示す(赤い線)。
そのレベルは-14.92eVであることが表示される。
キャンバス上でマウスをドラッグ(マウスボタンを押したまま左右上下に動かす)してみよう。分子が回転して表示されるだろう。
赤い球と青い球はP軌道を示している。回転させるとはっきりわかるが、この赤と青の球は色が逆転しているのが判るだろう。
これは位相が逆で節があるという。つまりパーオキサイドはHOMOが反結合性の軌道であることがわかる。分子軌道はHOMOまでは電子が詰まっている。
Upperボタンを押してみよう。そうするとLUMO(Lowest Unoccupied Molecular Orbital)が表示される。
LUMOには電子が詰まっておらず、空軌道であるという。
色々なエネルギー準位の分子軌道がどのような形をしているか確認してみよう。(注意:この分子軌道の形は速度をあげるため簡略化したものだ。単純に半透明の球を描いている。正しくは等高線図で書くので覚えておこう。)
この原料自体の過酸化物のデータは無かったが、例えば日油のカタログに次のような類縁体の過酸化物が記載されている。これを元に2012年講義で学生と計算を行ってみた。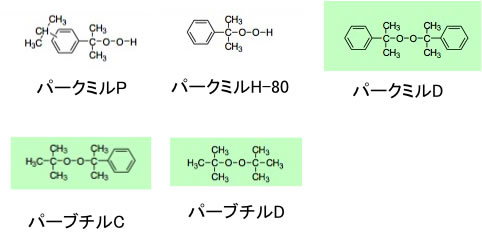
酸化物の危険の尺度としては10時間半減期温度で整理する事が多い。
10時間半減期温度というのは、その温度に保った時に、10時間で元の半分の濃度まで分解するという温度だ。例えば上記の過酸化物は次のようになる。
| 10時間半減期温度 | 活性化エネルギー | |
| パークミルP | 145.1 | 128.6 |
| パークミルH-80 | 157.9 | 125.6 |
| パークミルD | 116.4 | 158 |
| パーブチルC | 119.5 | 173.1 |
| パーブチルD | 123.7 | 155.8 |
この構造をYMBを使って分子のお絵描きを行い、様々な物性値を計算する。
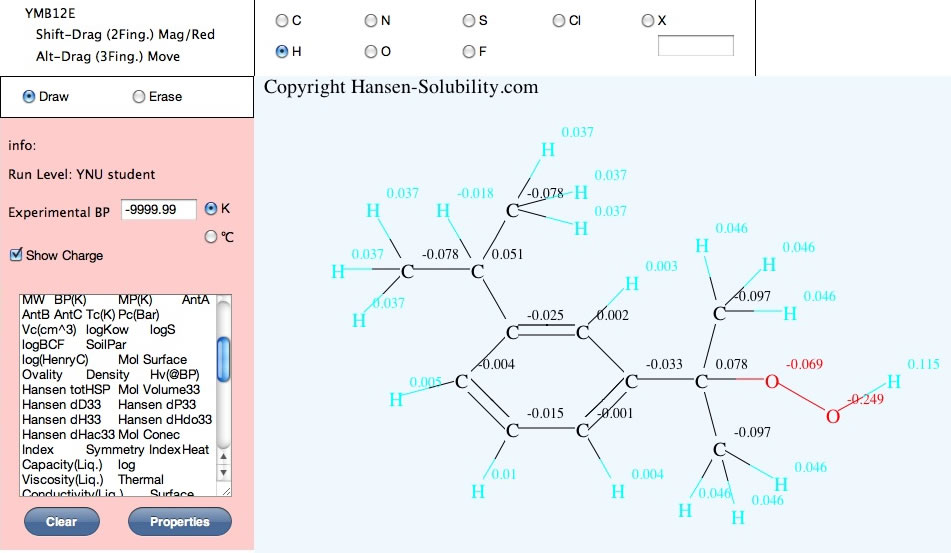
YMB12E(Chromeなどのブラウザー上で分子をお絵かきし、様々な物性を瞬間で計算するソフトウエアー)
企業からの訪問者はYMB12Eではなく、HSPiP に搭載のY-MBをお使いください。
そして、10時間半減期温度と、計算から出て来た物性値のうち、どの物性値(複数の場合もある)と相関が高いかを検討した。
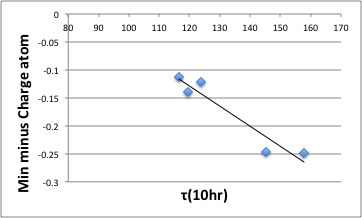
すると、原子上のマイナス電荷が大きいほど10時間半減期が高くなる事が示唆された。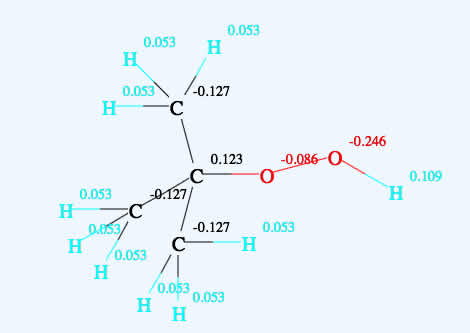
電荷はQeQ電荷平衡法を使って計算した。
後は、他の化合物で検証してみて(例えば上の化合物)、合わなかったら、他の識別子を導入するという操作を繰り返す。
さらにアシル化合物や、カーボネート化合物に拡張(後述)するなどして知識を深めて行く。
データというものは無限に増えていくので全てを学ぶのは無理だろう。
だが、データを還元して知識にすれば非常に少ない労力で様々なケースに対応できるようになる。
自分のやろうとするラジカル重合のモノマーの沸点、成長反応速度(ある以上の温度では重合しなくなる天井温度)、重合形態(バルク、溶液、乳化)によって最適な開始剤は常に異なる。
特徴を正しく理解すれば、(例えば電荷の反発が解裂の始まりなら)この反応は溶媒の種類に寄って(おそらく誘電率によって)分解速度が大きく異なるだろうと判断できるようになる。
実際に、ポリマーハンドブックにt-butyl peroxideの120℃での分解反応速度が記載されている。([2011年の講義参照-MOOC/Reactionりんくぎれ]())
| Solvent | Decomp. Rate *E-6 | Dielectric Cons. |
| carbon tetrachloride | 2.4 | 2.228 |
| toluene | 5.7 | 2.379 |
| triethylamine | 7.9 | 2.42 |
| dimethylaniline | 9.6 | 4.8114 |
| tetrahydrofuran | 9.7 | 7.58 |
| ethyl benzoate | 10.7 | 6.02 |
| 2-methyl-2-butanol | 12.6 | 5.82 |
| t-butanol | 14.1 | 12.47 |
| acetic acid | 21.9 | 6.15 |
| Acetonitrile | 22.1 | 37.5 |
これをプロットすると、大まかには分解反応速度定数は誘電率に依存していると言える。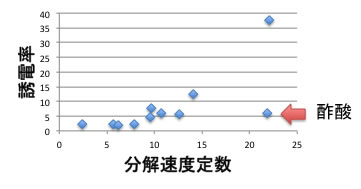
しかし、非常に大きく外れる酢酸の合理的な説明が無いと、知識を得たとは言えない。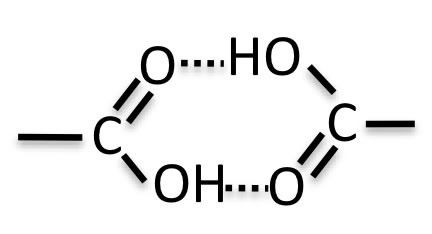
その理由は、酢酸は水素結合によってダイマーを作っているからだろう。本来カルボキシル基は非常に極性の高い化合物であるのだが、水素結合によって非極性の擬似分子になってしまい誘電率を下げてしまう。
その事は気液平衡の推算結果、蒸発潜熱と沸点の相関を表すTroutonの通則にも現れている。
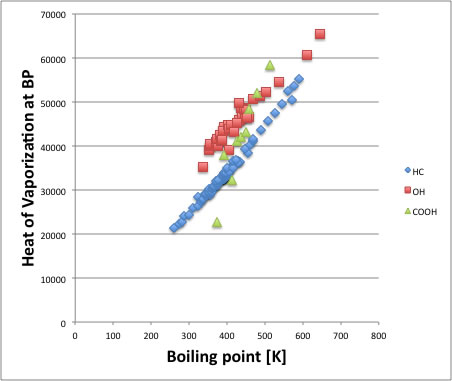
Troutonの通則をグラフにすると上図のようになる。炭化水素(HC)はほぼ直線になることが知られている。
アルコール(OH)は、HCの直線にほぼ平行で上側に来る。
ところが、カルボキシル基(COOH)を持ったものは、分子が小さいと、同じ沸点の炭化水素よりも蒸発潜熱が小さいことが知られている。
カルボキシル基の異常性は沸点が420K以下の(炭素数にして4ぐらい)のカルボキシル基を持ったものは、水素結合によって蒸発潜熱が非常に小さくなっていると理解されている。
そのような溶媒(アクリル酸やメタクリル酸も含まれる事に注意)はたとえ誘電率が低くても開始剤の分解反応は予想以上に早くなるだろうという知識が得られる。
| Carboxylic Acid | C# | Hv@BP | BP(K) | Hv@BP/BP | DiElectric Const. |
| formic acid | 1 | 59.5 | 58.5 | ||
| acetic acid | 2 | 60.6 | 6.15 | ||
| propanoic acid | 3 | 77.9 | 3.435 | ||
| acrylic acid | 3 | 67.8 | |||
| butyric acid | 4 | 81.9 | 2.97 | ||
| isobutyric acid | 4 | 79.4 | 2.73 | ||
| methacrylic acid | 4 | 82.9 | |||
| 3-methylbutanoic acid | 5 | 96.1 | 2.64 | ||
| pentanoic acid | 5 | 94.8 | 2.66 | ||
| 2-ethyl butyric acid | 6 | 104.1 | 2.72 | ||
| hexanoic acid | 6 | 94.0 | 2.63 | ||
| heptanoic acid | 7 | 109.6 | 2.5 | ||
| octanoic acid | 8 | 108.6 | 2.45 | ||
| Hexanoic acid, 2-ethyl- | 8 | 105.8 | 2.64 | ||
| nonanoic acid | 9 | 105.3 | 2.475 |
テーブルを埋めて、炭素数に対するHv@BP/BPに対してプロットしてみよう。
下のような図が得られるだろう。炭素数5以上では値が一定になることから、カルボキシル基は炭素数4以下ではダイマーを作っていると言われている。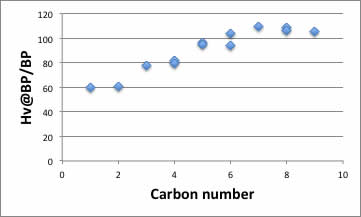
単純な炭化水素では、異性体を含めても下図のようになるので違いは明らかだろう。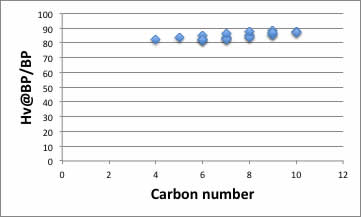
また、炭素数と誘電率をプロットすると以下の図になる。アクリル酸やメタクリル酸の誘電率は文献値が見つからなかったが、おおよその値は下図から求めることが出来るだろう。
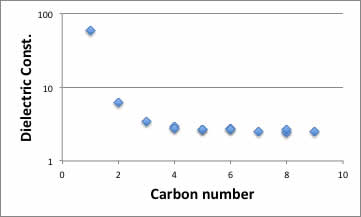
こうした検討を行うことが出来るのは、普段から蒸発潜熱などを利用して研究を行なっている化学工学系の研究者だけだろう。是非ともその優位性を発揮して欲しい。
有機化酸化物と並んでよく使われる、ラジカル重合用の開始剤に、アゾビス系の開始剤がある。
AIBNが最も著名であろう。
AIBNから生成するテトラメチルスクシノジニトリルはきわめて毒性が高いことが(許容濃度はシアン化水素の1/20)知られている。
そこで他の開始剤に置き換わりつつある。AIBNは以下の反応式で分解する。できたラジカルがモノマーと反応する前にカプリングをおこすと テトラメチルスクシノジニトリルになる。
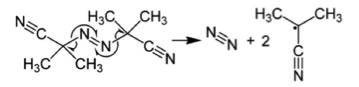
ここで、重要な概念、開始剤効率fというのを覚えておこう。
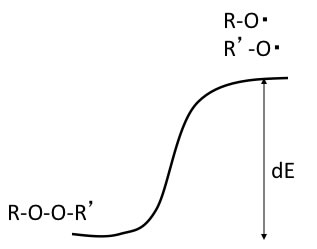
パーオキサイド化合物は熱がかかると坂を登って2分子に解裂する。
しかし、そのラジカルは2分子が反応して元のパーオキサイドに坂を転がり落ちて戻ることが出来る。
しかしアゾビス系の開始剤は真中の窒素が外れてしまうので生成したラジカルがカップリングすると、もう開始剤としては働くことができない。
そこで重合場に入れた開始剤のうちどれだけが有効に使われているかを示す開始剤効率fという尺度が非常に重要になる。
アゾビス系で0.6 – 0.7,パーオキサイド系で0.9 – 0.97あたりと言われている。
そこで問題になるのが、アシル系の開始剤だ。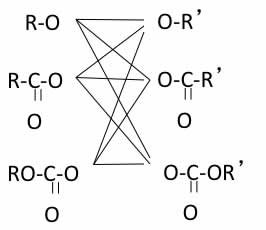
合成法上は任意の組み合わせの過酸化物を合成することが出来るのだろうが、アシル系の過酸化物はRによって壊れ方が異なる。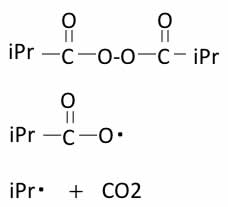
例えばRがイソプロピルのものは、10時間半減期温度が32.7℃と低く、分解しやすい。
反応を観察していると泡の発生が見られるため、炭酸ガスが出ていることがわかる。
しかしRがベンゼン環の場合、10時間半減期温度は73.6℃で、反応中泡の発生は殆ど見られない。
また合成されたポリマー中にエステルが導入されるので、NMRやIRで確認することができる。
つまり、過酸化物系でも開始剤効率は一律に高いのでは無く、アシル系(片方だけがアシルのものも含む)の開始剤効率は低い場合もあることを覚えておこう。
また、非対称の開始剤のうち下記のものなどは、メインのラジカル以外にイソブテンとt-ブタノールが生成する事がある。
こうした副反応はモノマーがガス状で溶解律速、高速反応で局所的にモノマーが不足(要はラジカル近傍に十分なモノマーがおらず、いつまでもラジカルのままでいるような時)したりすると無視できなくなる。
t-ブタノールの連鎖移動定数は0.46とメタノール、6.0と比べると小さいが高速反応を行なっている場合、もともと分子量は低くなる方向なので(後述)全体の物性バランスを取る際には注意が必要になる。
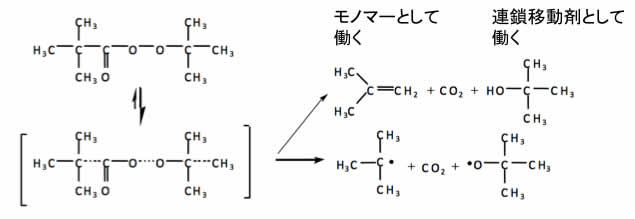
t-butyl peroxide( t-BuOOtBu )の場合も、生成したtBu・はアセトンとCH3・を生成する。
また、過酸化物の分解の場合は、生じたラジカルが誘発分解を引き起こすため、正確には1次反応には従わない。
アゾビス系の開始剤の溶媒依存性のデータがPolymer Handbookにある。4種類の溶媒に対する試験温度と分解速度の関係をプロットすると下図のようになる。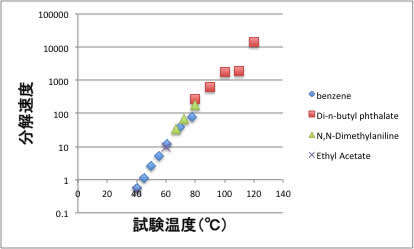
このように、一般的にアゾビス系では分解速度は温度だけに依存して溶媒の種類(誘電率)には依存しない。何故そのような結果になるのか考えてみよう。
このAIBNの分解速度が溶媒によらないという事実は、高分子関係の教科書には書かれているのは見たことがないが、溶剤ポケットブック(有機合成化学協会編、オーム社、昭和42年)では、溶媒効果の無い反応として説明されている。
10時間半減期温度
10時間半減期温度はものによって、溶媒効果を受けるので、カタログにある値を全ての系に適用はできない。
系特有の効果は温度の上げ下げで吸収できるとして、あるラジカル発生剤の経時変化を見てみよう。
ラジカル発生剤を100部使うと、10時間後には50部分解して、50部残る。次の10時間では25部分解して、25部残る。60時間反応しても3.125部残る。何時まで経っても0にはならないのだ。
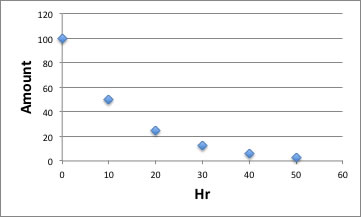
しかし、単位時間あたりのラジカル発生量はどんどん少なくなっていくことに注意しよう。
それは後にポリマーの成長反応の所で重要になる。
よくある間違いが、リアクターの利用効率をあげるため、開始剤をたくさん使って高速重合しようとする時だ。
(後に説明する生成ポリマーの分子量の問題、除熱を回避できたとしてもだ)
ラジカル発生剤の量を増やせば、ラジカルが多く発生し、単位時間あたりの収量も増える。
200部使えば、最初の10時間で100部が分解するので、前のように60時間かかっても97部しか分解しないのと比べ反応時間1/6になる。
と喜んでいるとポリマーの乾燥の時に真茶色になったり、再沈溶媒が異臭を放ったりする。
200部使えば10時間では、まだ100部未反応のラジカル発生剤が残っているのだ。
そうしたラジカル発生剤はその後も分解を続け、発生したラジカルは反応するモノマーがいないので、ポリマーや溶媒から水素を引き抜く。
水素を引きぬかれたポリマーや溶媒は再結合したり、転移したりするので期待していた性能が出なかったり、最悪廃液タンクが爆発するということも有りうる。
自分が設計したプロセス内でどれだけ開始剤が残っているのかはきちんと把握し、後処理で無害化する。
よく使われるのが、チオ硫酸ナトリウム(金魚用に使う水道水から塩素を無毒化する)だ。それにしても残存する開始剤量次第なので注意深く行おう。
反応熱
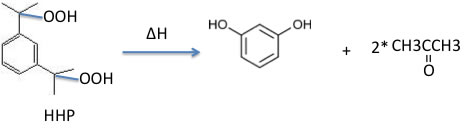
次に、YMBの計算結果のうち、Heat of Formationの値を使って、この反応の発熱量ΔHを求めてみた。工場では一日あたり20.8トン製造しており、そのタンクに保存されているHHPが全て分解したとしたらどのくらいの発熱量になるか計算した。
こうした計算が分子の絵を描くだけでできる(学生と実際に行ったが15分程度)ので非常に簡便である。1molあたりの発熱量は110.4kcalになった。
しかし、実際にはYMBは安定化合物を計算する為に作られたソフトなので、過酸化物の生成熱が正しく推算できているかどうかは明らかでは無い。
そこで、半経験的分子軌道法、MOPACを使って、これらの生成エネルギー(HF:Heat of Formation)を計算してみよう。
3次元構造の構築にはY-Molを使う。もっと簡単には。
Smiles構造式から3次元の分子構造を得るWebアプリ
JSME-RDKitから3次元の分子構造
(MOPACの計算環境の構築にはこちらの資料を参照すること)
HHP 生成熱 XXX kcal/mol
レゾルシノール 生成熱 XXX kcal/mol
アセトン 生成熱 XXX kcal/mol
ΔH = XXX kcal/mol
XXXを埋めよう。
分子軌道計算を行うにはもうひとつ理由がある。できるものがレゾルシノールやアセトンのように安定物質ならYMBで計算を行うことができるが、ラジカル重合のようにラジカルの生成熱を扱いたい場合にはYMBは全く無力だからだ。開始剤の生成熱(HF),ラジカルの生成熱を計算して下記のtableを埋めてみよう。
| 略語 | 10Hr半減期 | A | B | 開始剤HF | A-HF | B-HF | ΔHF | -CO2 |
| PBPV | 54.6 | tBuO | tBuCOO | 16.614 | -13.755 | |||
| PBA | 101.9 | tBuO | CH3COO | 17.479 | 9.987 | |||
| NPB | 73.6 | PhCOO | PhCOO | 17.502 | 17.198 | |||
| PHZ | 99.4 | PhCOO | PriPrO | 16.958 | 16.806 | |||
| PBZ | 104.3 | PhCOO | tBuO | 16.921 | 16.769 | |||
| PLIB | 32.7 | iPrCOO | iPrCOO | 17.626 | -29.726 | |||
| PBIB | 77.3 | tBuO | iPrCOO | 18.812 | -4.864 | |||
| PLMe | 67.9 | MeCOO | MeCOO | 17.200 | 2.217 | |||
| PLEt | 65.1 | EtCOO | EtCOO | 17.077 | -15.985 | |||
| PrCOO | 56.8 | PrCOO | PrCOO | 17.216 | -15.859 |
(A-HF+B-HF) – 開始剤のHFをΔHFに入れる。
脱炭酸する可能性のあるものは、A-HFを、R・とCO2として計算し、-CO2に結果を入れる。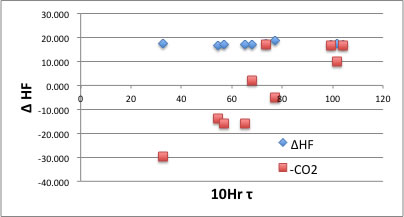
結果を10時間半減期温度に対してプロットし、どの開始剤が脱炭酸しにくいか判断してみよう。脱炭酸して安定化する開始剤は10時間半減期温度が低いのがわかるだろう。
開始剤の分解速度定数
通常の化学反応では、下図のような反応プロファイルをとる。
一般的には発熱量の大きいものは反応は進みやすい。
しかし、活性化エネルギーが非常に高ければ反応は進みにくい。この活性化エネルギーを求めるには、実験的には温度を変えた実験を数点行い、アレニウス・プロットをし、その傾きと切片から活性化エネルギーと頻度因子を計算する。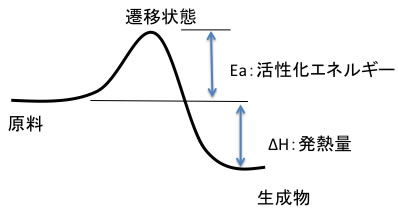
Kdを開始剤の分解速度定数、Aを頻度因子、Rをガス定数とすると、アーレニウス式は次のようになる。
kd=A・e^(- Ea / RT)
分子軌道法でも、ガウシアンを使い、基底関数をそれなりに高いものを使い遷移状態を求めれば、かなり精度よく活性化エネルギーを計算する事ができる。(藤本博、有機反応と軌道概念、化学同人、1986)
2分子反応の速度式は次のように書くことができる。(1分子の場合どういう式になるか確認しよう)

そこで、遷移状態を求め、エントロピー項(dS)とエンタルピー項(Ea)を求め、Kを計算してしまうことができる。MOPACの場合、分子の振動解析の結果があまり正しくないので、頻度因子は正しく求まらない。また、もともとラジカルの分解反応は、
のように、上に凸の山にはならない。そこで遷移状態の探索を行う(MOPACでTSというキーワードを使う)と多くの場合、計算は失敗する。
それは、遷移状態の探索が、「(山の頂上では)どの方向に動いてもエネルギーは低下する」構造を探索しているので見つからないという結論に達してしまうからだ。
(いくつか試してみたところ、うまく計算できたのは、tBuOOHだけだった。しかもその計算にはPM5以上の計算が必要なのでフリーのMOPACではダメかもしれない。)
それでは、計算しても無駄かというとそんな事もない。例えばフェニルパーオキサイド(PhPO)のTSを計算してみる。
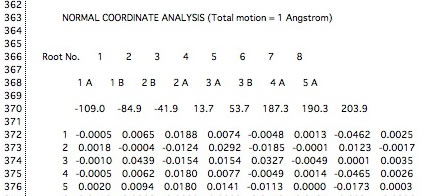
TSを求めた後、FORCEというキーワードを入れて振動解析をすると、虚の振動(-109.0, -84.9, -41.9)が3つあるのでこれは遷移状態ではない。
(正しい遷移状態では、虚の振動は1つで、その振動方向が反応の進行(逆進行)方向である必要がある。)
しかし、その-109.0の振動を実際にみてみると、次のようになっていることがわかる。
これを見た時に何を感じるかは、研究者の直感次第だろう。自分は、こうしてねじれながら離れていくなら、マイクロウエーブをかけながら重合したらPhPOはどうなるか非常に興味を覚える。(パーオキサイドの実験は非常に危険を伴う。責任は負わないので、実際にやるならリスクアセスメントをきちんとやるように。)
分解反応における圧力効果
エチレンやプロピレンの重合を扱っていない限り、超高圧で重合を行うことはまれだろう。様々な触媒が開発されているので超高圧重合の知見は失われて、開始剤の分解反応の圧力依存性に関する項目は高分子化学の教科書から失われているように思える。
自分の持っている本の中では唯一「高圧化学反応」(K. E. Weale著、蒔田薫ら訳、培風館、1969)に記載がある。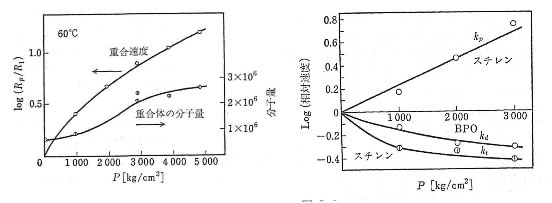
系はスチレンで、開始剤はBPOを用いている。
圧力が高くなると、重合速度は(logプロットなのに注意)非常に高くなるが、分子量の増加は僅かである。
重合は2分子の衝突によって起こるので、圧力の増加は反応を加速する。
しかし、開始剤は逆に、1分子が2分子に解裂する反応なので、圧力の効果は逆に働く。
単位時間あたりのラジカル発生量が少なくなれば、その分、分子量は高くなる。スチレンの停止反応は再結合(後述)が主なので、圧力によって増加するはずである。
しかし、高分子同士が再結合するためには、高分子末端のラジカルが出会う必要があり、これは拡散の影響を強く受ける。そこで圧力の効果は相殺され(というか逆に圧力が高いとktは小さくなる)右図のような反応速度ー圧力依存になる。
通常のラジカル重合ではそのような超高圧を考える必要は無いが、圧力が高いと開始剤は分解しにくくなるとおぼえておこう。(超臨界CO2重合をやる場合には思い出そう)
開始剤の反応速度論
開始剤の反応速度論をまとめておこう。
Kd:分解速度定数、温度や溶媒(誘電率?)に依存する。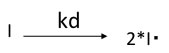
開始剤の分解の反応速度、Vd=kd[I] となる。[I]:開始剤濃度
パーオキサイドなどの開始剤は1分子から2個のラジカルを生成するので、ラジカルの生成速度は2kd[I]になる。生成したラジカルがモノマー(M1)に付加するとM1ラジカルができる。
モノマーラジカルM1・の生成する開始反応速度viは
vi=d[M1・]/dt = 2fkd[I]
f:開始剤効率
ラジカル重合
開始反応
成長反応
連鎖移動反応
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください)
メールの件名は[pirika]で始めてください。