
| ホームページ | Pirikaで化学 | ブログ | 業務リスト | お問い合わせ |
| Pirikaで化学トップ | 情報化学+教育 | HSP | 化学全般 |
| 情報化学+教育トップ | 情報化学 | MAGICIAN | MOOC | プログラミング |
2021.4.4
日本を代表する車メーカーから蒸気圧の推算に関して相談のメールをもらったことがあります。
質問:
ガソリンは、300種類を超える成分の混合物ですが、各々の成分の沸点、蒸気圧は推算可能でしょうか?
今回はこの質問を考えてみましょう。
まず、炭水化物だけを考えましょう。
炭水化物というのは、炭素と水素だけからできている化合物のことです。
その定義ですと、ベンゼン、トルエンなどの芳香族化合物も炭水化物になるのですが、多くの場合、芳香族は別に考えます。(ガソリンと括る場合、本来はデータに混ぜなくてはなりませんが今回は割愛します。)
まず、異性体について学びましょう。
枝分かれ、2重, 3重結合、環、cis/transなどを考慮に入れてください。
(アキシャル/エクアトリアル、光学異性体は考えないことにしましょう。)
10種類以上、 20種類以上、30種類以上、40種類以上。
「どれだと思う?」と聞くと、大抵の場合、4択の上から2番目の30種類以上に手を上げる学生が圧倒的に多いです。
電気製品とかを購入する時の行動に似ているような気がします。
答えを見る前に、どのように考えれば網羅的に答えを得られるか考えましょう。
"考えましょう”と言うと、”わかりません。ちゃんと覚えますから答えを教えてください”と言う学生が多いです。
覚えるだけでは、人間はAIにはもう敵いません。
記憶力だけを頼りに受験戦争を勝ち抜いて来て、大学生になっても”考える力”を磨いてこなかったのなら、もう諦めてAIに頼りましょう。
本当にちゃんと考えて紙に全部書いてみた?(▶︎をクリックして開く)
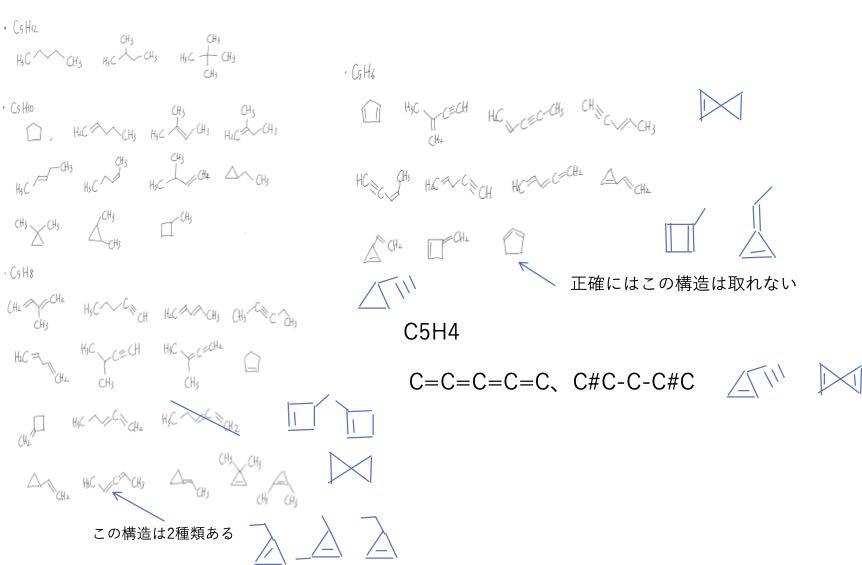
戦略的には、C5H12からHを2つ減らしながら構造を考える。
環は1つあたり2つHが減る。
2重結合は2つ、3重結合は4つ水素が減る。
この学生はその戦略はできていたけど、全てを数え上げることはできなかった。
私がいくつか書き足しているけど漏れがないか自信を持って言えない。
全くもって、いい加減な先生だ。。。。
答えが正しいかどうかではなく、戦略があっていたかどうかを確認したら次に進みましょう。
このように、C5Hxですらこのように大量の異性体が存在するので、ガソリンの主成分、イソオクタン(C8Hx)では飛躍的に異性体が増えることは容易に予想できるでしょう。
そうしたC8近辺の異性体の蒸気圧を知りたいと言うのが、T社の質問者の聞いて来たことです。
この時には、C8H18から順番に水素を2つ減らし、構造を考えると言う戦略は少し難しいかもしれません。
それでは、どのように考えるのかと言うと、炭素のとりうる状態を全て書き出します。
炭素の混成軌道は、SP3, SP2, SPの3種類あります。それに水素を付加した官能基がいくつあるか数え出してみましょう。
考えもせず開いてはいけません。。。(▶︎をクリックして開く)
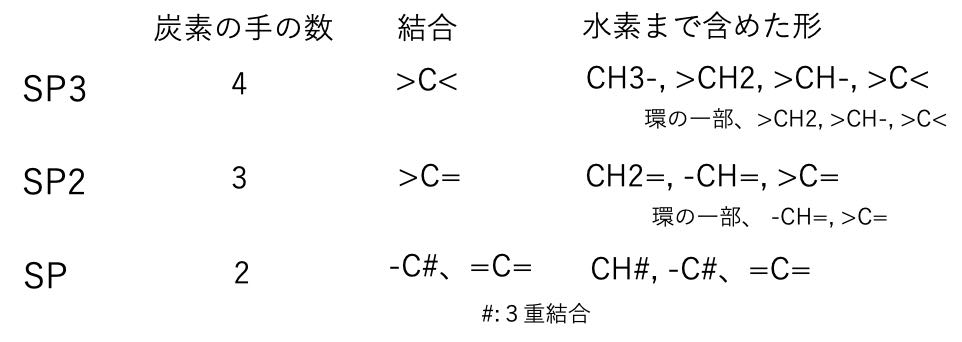 つまり、これらの官能基を8つ選んで、組み立てることができる化合物が何種類あるか?と言う問題になります。
つまり、これらの官能基を8つ選んで、組み立てることができる化合物が何種類あるか?と言う問題になります。
コンピュータを使って、片っ端から構造を作成するのでしたらこのやり方がベースになります。
課題:
ループを回すと、同じ化合物が生成されてしまうことがあります。例えば左から見た場合と右から見た場合で同じ構造であることがあります。
プログラムでそれを処理するのは非常に大変です。
コンピュータは幾何学は不得意です。
(これは正確な言い方ではありません。人間が幾何学的なプログラムを書くのが不得意なだけです。)
重なりも含めて全て書き出し、同じ化合物を除外する方法を考えてみてください。
このような官能基が何個あるかで物性を予測する方法として、Jobackの原子団寄与法と言うものがあります。こちらでこの方法を詳しく説明しています。
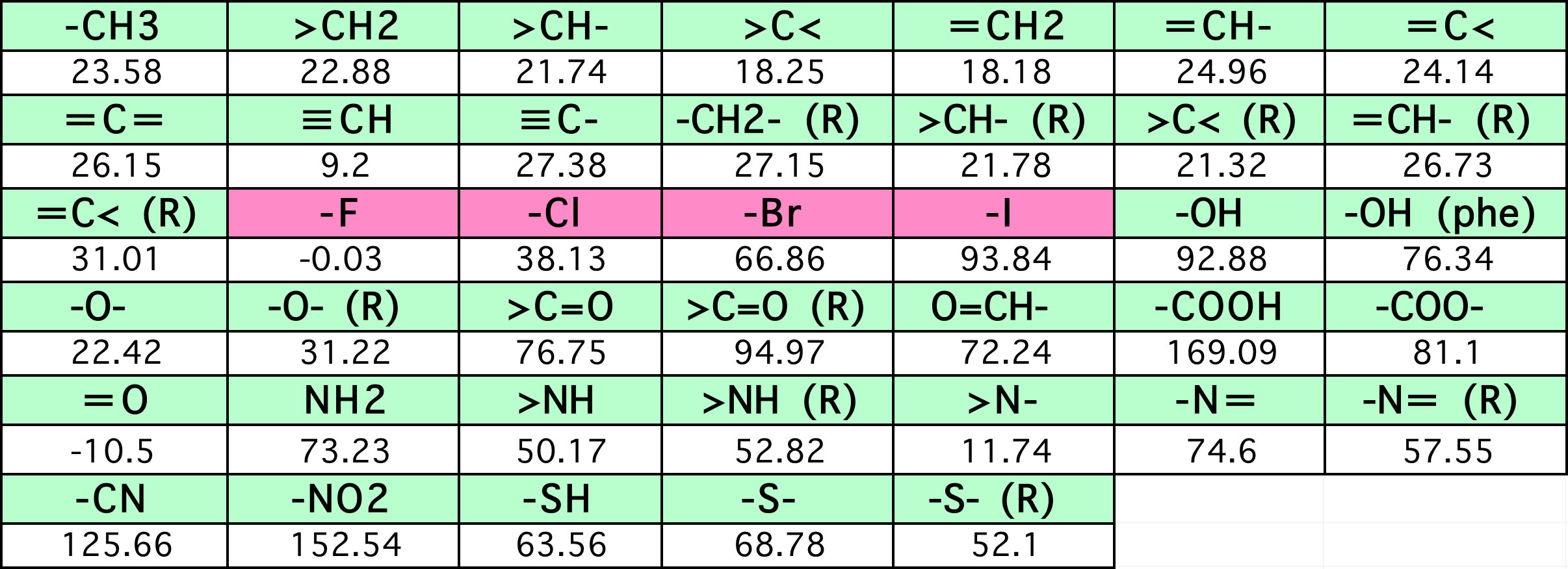
沸点=198.2+Σ(原子団の数 * 表中の係数)
そこで、同じように、原子団の数から蒸気圧を計算するのに必要なパラメータを予測する式を作ってしまえば良いことがわかります。
蒸気圧を予測する式にどんなものがあるか?はこちらにまとめてあるので参照してください。
ここでは、Antoine定数を使うことにします。
Ln(P[mmHg])=A-B/(T[K]+C)
実際にはlog,lnであったり、mmHg,kPaであったり、℃,Kであったりするので、データ集によって値は様々です。
この、A, B, Cの3つの値は化合物特有の値になります。
Antoine 定数のJoback原子団寄与率を定めてみましょう。
データソースはJoback先生の名前を広く世界に知らしめた、The Properties of Gases&Liquidsに収録のデータを使います。
余り、広げすぎても大変なので、芳香族を除く炭化水素化合物に限ります。
書籍に記載のデータ439化合物のうち、161化合物が該当します。
一覧を表計算ソフトにペーストしておきましょう。
次に、各化合物がどの原子団を何個持っているかの一覧表を作ります。
CH3 CH2 CH C CH2= CH= C= .=C= #CH #C CH2_R CH_R C_R CH=_R C=_R
手作業でやりますか?
これぐらいならどうにかなるかもしれませんが、他の官能基が入って来ても、間違いなく作れるか想像してみましょう。
想像もせず開いてはいけません。。。(▶︎をクリックして開く)
原子団の取り方は、Joback先生はJoback法の定義を使いますし、PirikaはPirikaの分割法をとります。
原子団への分割を間違えたら、意図した推算結果になりません。
推算式を作ってみたら精度が出ないので、新しい原子団を追加したくなることもあります。
その度に原子団のテーブルを作るのは非常に効率が悪いのです。
例えば、今回の分割法を変更して、エチル基(CH3CH2-)を定義したとして、間違いなく全部修正できるかは大問題になります。
そこでpirikaでは、原子団のテーブルを持たせると言う戦略を捨てて、Smilesの構造式だけを持たせることにしました。
そして、Smilesの構造式を中間コードに変換して、任意の方法の原子団セットに変換するプログラムを作成しました。
ユーザーに徹するなら必要ではないかもしれませんが、MAGICIANを目指すのであれば、プログラミングも必須です。
炭化水素化合物は単純で余りおかしな値を持つものは多くありませんが、一般的にAntoine定数は3つの値の非線形式になるので、同じパーフォーマンスを示す3つの数字の組みは無数に存在します。 したがって原子団寄与率がどのような系でも求まると思うのは間違いです。 今回、これができるのは、炭化水素だけを相手にしているからです。
AntoineのA,B,Cパラメータと原子団のテーブルが準備できたら、後はいつものようにYSBを使って解析を行うだけです。使い方を忘れていたら、こちらを読み直しましょう。
でも、その前に変なデータを除去したほうが良いです。
(データのクレンジングと言う人も多いですけど、クレンジングと言うのは”化粧落とし”と言う意味の和製英語です。洗い清める(cleanse)にingつけたのでしょうね。クレンザー(cleanser)が研磨剤入りの汚れ落としなので違和感はありませんが。)
簡単のため、沸点に対してA,B,Cパラメータをプロットして、おかしな値を除去しておきます。
表計算ソフトの側でA,B,Cパラメータを持たない化合物を除去したテーブルをYSBを使って解析を行います。
これから先は、どのデータを除外して計算を行ったかによって結果は変わります。
まず、重回帰の結果です。(YSBのクロスターム=0に相当します。)
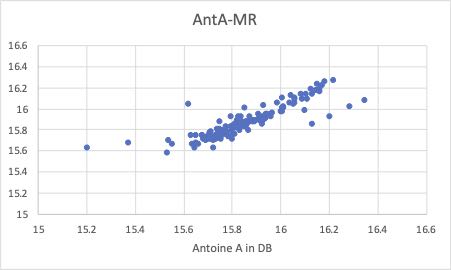
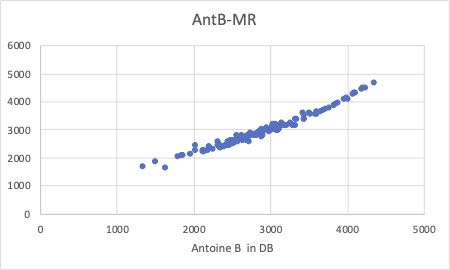
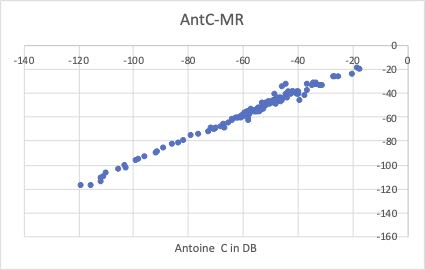
特にAntoine Aの値で、除去したくなる誘惑に駆られるデータがいくつかあります。
Antoine定数は、3つのペアで意味をなす値なので、Aの値が悪くても他のB,Cで全体としては調整されていることもあります。しかし、ある特有の非線形性相互作用があってずれることもあります。
単純に合わないものを消すのではなく、合わないものを特定して、吟味して、どうするか決めていく必要があります。
私がどちらかと言うと問題に感じるのは、Antoine Bです。屈曲点を持っているように見えます。(値が大きい所と小さいところで傾きが異なる。)
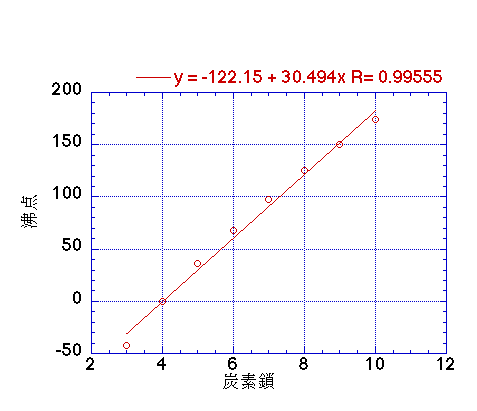
直鎖のアルカン化合物の炭素数を沸点に対してプロットするとこのようになります。
狭い範囲では直線ですが、大きくなるにつれて寝て来ます。
つまり、JobackのCH2パラメータは大きくなるにつれ線形ではなくなると言うことです。
そこで、クロスターム重回帰法を使って解析してみます。
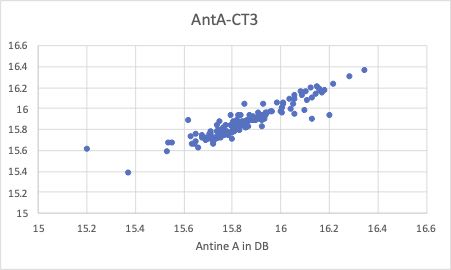
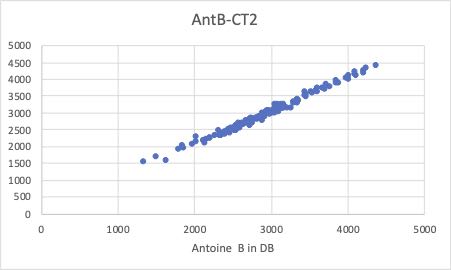
Antoine Bパラメータで、屈曲がなくなっていることがわかると思います。
どのようなクロスタームが選ばれたか見てみると、-7.06872137580808*CH2*CH2が最初のクロスタームになります。
これが何を意味するのかと言うと、CH2に関しては線形ではなく、上に凸の非線形で考えるべきだとYSBが提案していることになります。
次のクロスタームは、-10.9176863572035*CH2*CH2(環)です。
つまり、分子の主骨格を作るCH2構造が増えるとAntine Bは基本大きくなるのですが、だんだん、その効果は小さくなっていくのを、このクロスタームは示しています。
Antoine Aの場合、クロスタームを3つ、Antoine Cの場合クロスターム0で良しとしましょう。Antoine Bをクロスターム0と2とした時でどう変わるか評価してみましょう。
大事なことは、分子の構造が決まれば(テーブル中の官能基の数が決まれば)Antoine A, B, Cの予測値が求まると言うことです。 A,B,Cパラメータを持たない化合物も沸点のデータはあるので、検証に用います。
Ln(P[mmHg])=A-B/(T[K]+C)
P=760mmHgになる時の温度、T[k}は次式で求まります。
T[k]=B/(A-LN(760))-C
この時のBをクロスターム0と2で温度の推算値がどう変わるかみてみましょう。
まず、屈曲点がある、Antoine B クロスターム0の場合です。
(Antoine A はCT=3, Antoine CはCT=0の推算値を使います。)
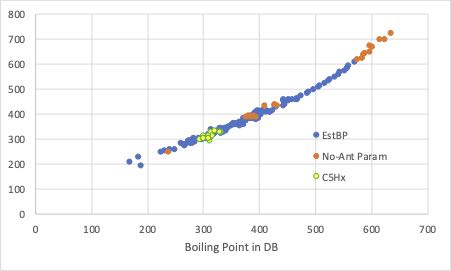
Gases&LiquidsにAntoine 定数が未記載のものはオレンジ、Gases&Liquidsには存在しないC5Hxの化合物でPirikaのDBに沸点のあるものについては黄色でプロットしてあります。
C5Hxの化合物のリストは以下のようになります。
官能基のリストを自分で完成させて、Antoine A, B, Cを予測し、DB中の沸点と比較してみましょう。
結果を見ると、沸点は良好に推算できているので、Anoine A, B, Cはそこそこ正しく予測できているようです。
ただし、Antoine Bと同じように、沸点に関しても同じように屈曲点が現れます。
沸点が500K以下であるなら沸点の予測精度はとても高いと言えます。
それに対して、Antoine Bの推算に、クロスタームを2つ導入し、屈曲点を消したものを使って沸点を計算してみます。
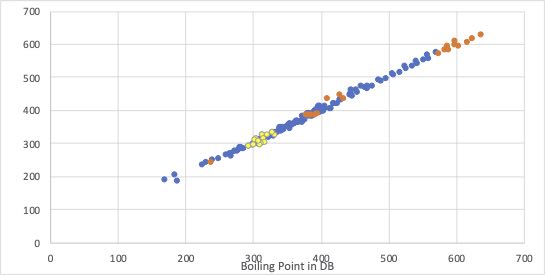
今度は屈曲点が消えて、全域で高い推算精度を持っていることが分かります。
このように、T社の研究者からの問い合わせに関しては、芳香族を除く炭化水素化合物に関して、任意の構造の化合物の任意の温度での蒸気圧はこのぐらいの精度で推算可能と答えることができます。
課題:
現実には、Gases&Liquidsのデータの中には、C=_Rを使った化合物が1つもありませんでした。そこで、このパラメータを決めることができません。
どうやったらそうしたデータのない原子団の寄与率を決定できるか考えましょう。
考えもせず開いてはいけません。。。(▶︎をクリックして開く)
原子団寄与法は比較的精度が高いモデル式が構築できますが、どんどん原子団が増えるので、データが追いつかない事がよくあります。沸点を推算するために原子団を導入したのは良いけど、沸点以外のデータは全くないなどは良くあります。
そうした時には、説明変数のセットを変えます。
例えば、蒸気圧の推算で説明したように、臨界定数を用いて対応状態原理を用いて蒸気圧を推算するのも良いでしょう。
YMBはAntoine定数を推算するので、わざわざ臨界定数をYMBで推算して蒸気圧を計算するのは本末転倒ですが、練習になりますのでトライしてください。
対応状態原理で、C=_Rを使った化合物の蒸気圧曲線を求め、Antoine定数を仮決めします。将来実験値が出てきたら差し替えるのです。
また、全く系列の異なる式を立てるのも有効です。
RDKitはトポロジカルな識別子をSmilesの式から作成する事ができます。
試しに、変数選択重回帰法を使って変数を選び、Antoine A, B, Cを推算して沸点を予測してみました。
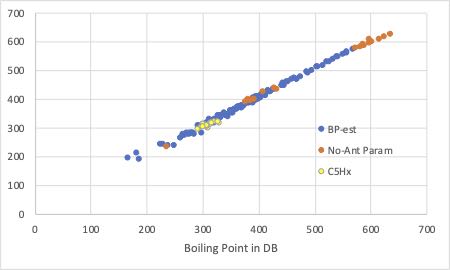
自分で、どんな識別子を選択したらこのような結果が得られるか試してみてください。
自分で作った式が満足いくものであったら、RDKitから作成したAntoine定数からC=_Rのパラメータを決めます。
さらに、RDKitは3次元の分子構造も吐き出してくれるので、分子軌道計算を行い、その結果からAntoine定数の予測式を作ることもできるでしょう。
このように様々な種類のモデル式から作成した、C=_Rの寄与率がみな似ていれば、その値を安心して使えると言うことです。
原子団寄与法は、分子を原子団にバラバラにしてしまうので、結合情報は失われてしまいます。例えば、cis体とtrans体はバラバラにしたら同じになります。
トポロジカルな識別子を併用する方法が大事にあるでしょう。
さて、それでは、T社の研究者は何を根拠にガソリンは300種類以上の混合物というのでしょうか?
多くの場合は、混合物の中の成分を分析するには、ガスクロマトグラフィー(GC)を使います。昔はシリカの微粉末をカラムに詰め、シリカと化合物の相互作用の大小で分離していました。最近は、キャピラリーカラムという、液膜との相互作用の大小で分離する事が多くなってきたと思います。
検出器は、TCDやFIDを使います。(私は詳しくないので自分で調べてください)
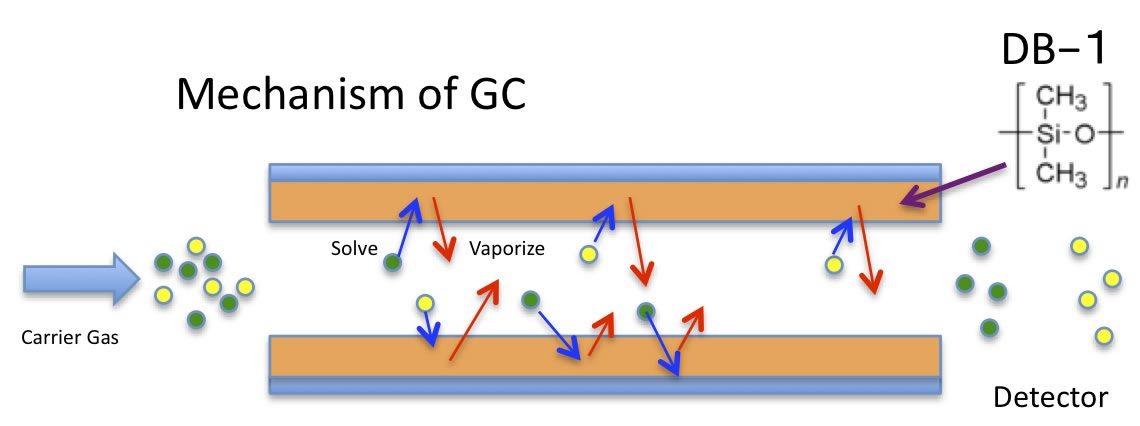
このGCでガソリンを分析すると、ピークが300個以上現れる事を根拠にしているのだと思います。
ガソリンの主成分がイソオクタンだとして、似たような沸点の化合物が無数にあります。そこで原油を蒸留しても、似た沸点のものは蒸留では分離できずに混入してしまいます。
このガスクロ分析は、カラムの長さやオーブンの温度などによって、保持時間(リテンションタイム)は異なります。そこで、保持時間の絶対値は意味を成しません。
そこで、KovatsのRetention Indexというものを使います。これはノルマルのアルカンの保持時間を、炭素数*100で規格化する方法です。
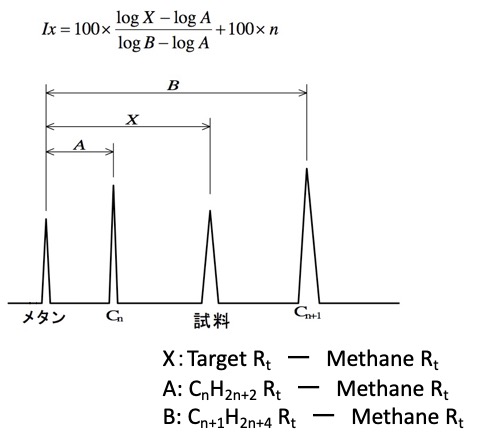
このGCRI(ガスクロのリテンション・インデックス)のデータベースはNISTがまとめています。そこから、芳香族を除く炭化水素のデータをとってくると以下のようになります。
課題:
GCRIの予測式を作成し、データのない化合物のGCRIを予測してください。
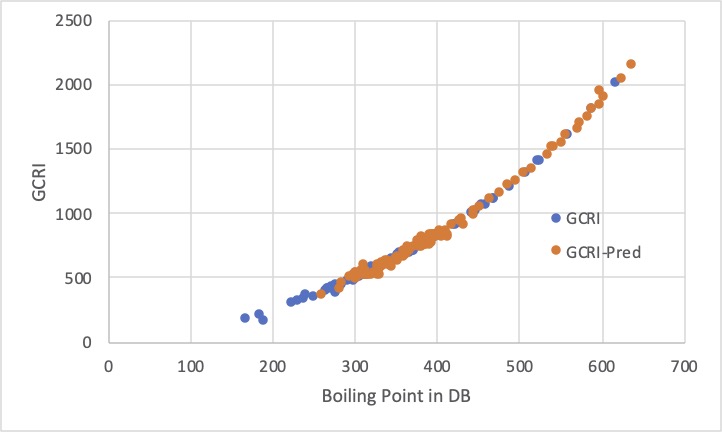
試しに私がRDKitの識別子から作ってみたところこのようになりました。
そこそこの精度ような気がしますが、用途によって良く考えましょう。
例えば、GCRIが500付近にある化合物は、沸点にすると290-325℃付近になります。
そのぐらいの沸点の化合物は全て、GCRI=500と予測されてしまうのでしたら、ガスクロで分離できないという答えになります。
その辺りの精度を上げたいのであれば、RDKitの識別子の選び方をどう変えたら良いか?
解析用プログラムを自分で作成できないなら、どう工夫したらそれが実現できるか考える。
それを考える事なしに、データをこね回していても社会の要請には答えられないという事です。
最後に気分を変えてニューラルネットワークがどのように沸点を学習するかまとめてありますので、自分で教育してみてください。
Copyright pirika.com since 1999-
Mail: yamahiroXpirika.com (Xを@に置き換えてください) メールの件名は[pirika]で始めてください。