Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
Chapter 22, Chromatography – HSP creator and user (Retention Times and HSP)
One important aspect of chromatography is
as a creator of reliable HSP. Given how hard it can be to get HSP for chemicals
and polymers, it’s good to know that those with the right chromatographic kit
can determine HSP relatively easily.
HSP can also be used to explain and, more
importantly, predict retention times in some chromatographic processes. So
chromatography can be both a creator and user of HSP.
Creating
HSP with chromatography
The archetypal method is Inverse-phase Gas
Chromatography (IGC) where the solid phase is made from the polymer to be
investigated and the retention times are measured for a series of solvents with
known HSP. The closer in HSP the solvent is to the solid phase, the more the
solvent will tend to linger within the solid phase, so the retention time will
be higher. However, as we will see, IGC also depends on other factors such as
vapour pressure.
The key issue is how to extract HSP from
the retention data. The first step is to convert the retention times into
specific retention volumes Vg. These can then be converted into Chi parameters.
Finally, the HSP can be found by doing a linear regression fit to the formula
relating Chi to HSP. The papers of Professor Jan-Chan Huang’s group at U.
Massachusetts-Lowell are good examples of this, J-C Huang, K-T Lin, R. D.
Deanin, Three-Dimensional Solubility
Parameters of Poly(ε-Caprolactone), Journal of Applied Polymer Science,
100, 2002–2009, 2006. Their data can be recast into a format that uses
the standard HSP distance (in this case, distance-squared). The famous factor
of 4 is automatically included. Careful analysis by Professor Huang’s group
found that by having no factor of 4
the errors were rather large and they included a factor as a fitting variable.
Its optimum values varied from 2.6 to 4. The group were very much aware of the
theoretical value of 4 but postulate that at the higher temperatures of these
experiments the lower values may be justified.
Here is the fit for their data on
polycaprolactone, using the revised version of their approach
Equ. 1‑1 Chi = C1 + MVol *C2 * (4 * (δDp-δDs)2
+ (δPp-δPs)2 + (δHp-δHs)2)
Where constants C1, C2 and the Polymer HSP
(δDp, δPp, δHp) were the fitting parameters.
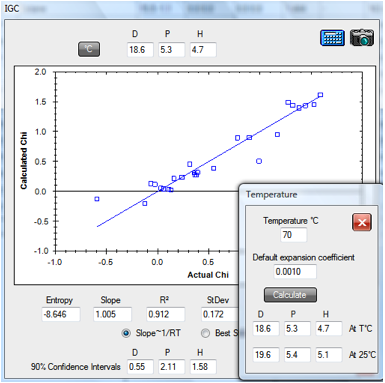
1‑1 IGC fit of polycaprolactone data
From this fit, the values [18.6, 5.3, 4.7]
are close to Huang’s optimized fit (using 3.2 instead of the factor of 4) of [18.5,
4.2, 5.6]. The fitting program contains extra information based on Huang’s
analysis. An Entropy value captures the size of the RTη term (using an
approximation of 100 for the Molar Volume of the stationary phase). And a 90%
Confidence Interval is estimated for δD, δP and δH using an approximation to
the method described in Huang’s paper.
The program offers a choice of fits to give
the user some idea of the robustness of the results. The “C2*RT ~1” option
forces C2 to be close to its theoretical value of 1/RT. Alternatively you can
optimize to the minimum Standard Deviation. You can also explore the robustness
of the fit manually be entering your own values into the δD, δP and δH fields.
Note that the Temperature (°C) option is
used so that solvent HSP are calculated at the 70°C of the dataset as discussed
below. When translated back to 25ºC the values become [19.6, 5.4, 5.1].
The dataset used (Polycaprolactone Huang
Probes.hsd) is provided with HSPiP along with a fuller dataset
(Polycaprolaction.hsd) which gives a fit of [19.4, 5.5, 5.6].
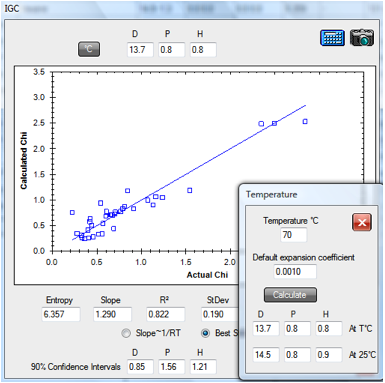
Similarly the 70ºC PDMS data set
(PDMS.hsd) gives a room temperature value of [14.5, 0.8, 0.9].
Figure 1‑2 IGC fit of PDMS data
Huang’s analysis is of data from M. Tian
and P. Munk, Characterization of
Polymer-Solvent Interactions and Their Temperature Dependence Using Inverse Gas
Chromatography, J. Chem. Eng. Data 39, 742-755, 1994. HSD files for all the
polymers in this paper are included with the HSPiP as a contribution to the use
of IGC for HSP purposes. Note that the Chi parameters are calculated in the
Tian and Munk paper. See below for doing your own Chi calculations.
If that were all there is to it then
everyone would be using IGC to measure HSP of their polymers. One issue is that
IGC has to take place at a temperature where the polymer is suitably “liquid”.
In the example above, the HSP for the polymer are those at 70ºC. The
fitting procedure compares the polymer HSP with the solvent HSP. But of course,
the validity of the calculation depends on having good values for the HSP of
the solvents at these elevated temperatures. The standard approximation for
calculating HSP at different temperatures comes from Hansen and Beerbower:
Equ. 1‑2 d/dT δD = -1.25a δD
Equ. 1‑3 d/dT δP = - a δP/2
Equ. 1‑4 d/dT δH = -(1.22 x 10-3 + a/2)δH
a is the thermal expansion coefficient. For the IGC technique to become
more popular we need ready access to reliable calculations of HSP at different
temperatures for the common solvents so that data from different IGC users will
be comparable. HSPiP includes those calculations based, wherever possible, on
known temperature dependent as. The full Sphere Solvent Data set contains the appropriate
coefficients for calculating a
at a given temperature from the constants a, m and Tc
(the critical temperature):
Equ. 1‑5 a = a*(1-T/Tc)m
Note that this equation is only valid over
a defined temperature range. For solvents with no constants or outside their
valid range, HSPiP uses a user defined default value for a.
However, many molecules which are
adequately “liquid” at room temperature can be used as the “solid” phase for
IGC. The paper by K. Adamskaa,, R. Bellinghausen, A. Voelkel, New procedure for the determination of
Hansen solubility parameters by means of inverse gas chromatography, J.
Chromatography A, 1195, 2008, 146–149 measures the HSP of a group of
excipients: Cetiol B, Lauroglycol FFC, Labrasol, Miglycol and Tween 80. They
used a similar fitting procedure for deriving the HSP. Professor Voelkel kindly
sent the Chi data for Tween 80 and here is an Excel fit of their data:
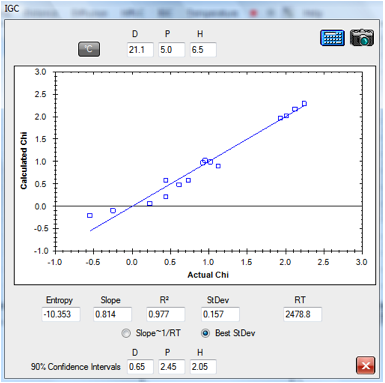
1‑3 IGC fit of Tween 80 data
The fitted HSP for Tween 80 [21.1, 5, 6.5]
gives a suspiciously high δD value – for reasons that are unclear at this
early stage of the science of interpreting these data.
The other issue is that the calculation of Chi
from Vg requires knowledge of the partial pressure of each solvent at that
temperature plus the second virial coefficient. This is an important point. If
we measured the retention time of a new solvent on a range of IGC columns, we
could do exactly the same calculation to determine the HSP of that solvent. But
how many of us know the partial pressure and second virial coefficients of our
new solvents?
As an aid to the IGC community, and as an
attempt to provide a common standard for these tricky calculations, a
calculator for converting specific retention volumes, Vg, to Chi parameters is
provided. A representative range of solvents is provided, spanning a good range
of HSP space. For each solvent Tc, Pc and Vc (the three Critical Parameters)
and Antoine A, B, C parameters are provided. These in turn are used to
calculate P0 (the saturated vapour pressure) of the probe solvent
and B11, the Second Virial Coefficient. It turns out that there are
a number of alternative methods for estimating B11 from the Critical
Parameters, and they give different results. Happily, the exact value of B11
is not hugely important so we don’t have to worry too much. To help users, two
representative methods are used. The first has been popularised by Voelkel and
uses Tc and Vc. The second is coded BNL after Blu, Nesterov and Lipatov, which
uses Tc and Pc. In general the BNL method gives larger (more negative) B11
values than the Voelkel method. The program shows you the B11 values
(and P0) so you can see for yourself. You can find out all about
these methods (and others) from A. Voelkel,, J. Fall, Influence of prediction method of the second virial coefficient on
inverse gas chromatographic parameters, Journal of Chromatography A, 721, 139-145,
1995. The only other inputs are the density of your test material and its
“molar volume”. Both values are controversial. The density, of course, changes
with temperature. But as the molar volume of the solvents also change with
temperature (and are not explicitly calculated) the assumption is made that the
relatively small density effects cancel out. The “molar volume” is often an
estimate – what, for example, is the molar volume of a polymer? The
recommended guess is to put in the molecular weight of a typical monomer unit.
Errors in this parameter mostly result in an offset of the calculated Chi
values rather than a significant change in their relative values. The
calculation of HSP is relatively insensitive to this offset. Because you can
instantly see the effects of varying the density and the “molar volume” you can
make your own judgement of how valid the calculated HSP really are. It’s important to note that these sorts of
judgements about B11, density and molar volumes are generally not explored in
IGC papers. By making them explicit, and by encouraging the use of the same
basic dataset, we hope that the IGC community will start to standardise on one
approach so that calculated HSP are relatively more meaningful.
So although IGC has considerable promise
for determining HSP of polymers and
looks excellent for intermediate molecules such as excipients, oligomers and
surfactants at room temperature, its use for taking a set of known stationary
phases and deducing HSP of solvents looks like something that is not for the
faint-hearted.
There’s one key point about IGC. It’s
relatively easy to get good fits from just about any data set. But the value of
that fit is questionable unless the test solvents cover a wide region of HSP
space. This is the same issue as other HSP fitting. Having lots of data points
in one small region is far less valuable than having the same number of data
points spread out over the full HSP space. One IGC paper used 5 similar alkanes,
2 similar aromatics, 3 chlorinated hydrocarbons and one simple ester. This is a
very small portion of HSP space! So if you want really good IGC data make sure
you challenge the material with solvents from the full range of alkanes,
aromatics, ketones, alcohols, esters, amines etc. etc. The wider the range, the
more likely it will be that your calculated HSP are meaningful. That’s why in
the Vg to Chi calculator we’ve included not only solvents typically found in
IGC papers but a few more that will help cover a broader HSP range. By
providing a consistent set of Antoine and Critical Parameter values we will
have saved IGC users a lot of work!
GC
prediction via GCRI – Gas Chromatography Retention Index
It would be very useful if we could predict
in advance the exact retention time of a chemical on a GC column. But of course
this is not directly possible as retention time depends on numerous specific
variables in the GC analysis: column type, column dimensions, temperatures,
flow rates etc.
A useful substitute is the GCRI. We know
that the retention time of straight chain alkanes form an orderly progression
from CH4 to C2H6 to C3H8 … And if we give each of these alkanes a retention
index = 100 x number-of-carbon-atoms we can say, for example, that if a
chemical elutes at precisely the same time as heptane then its GCRI is 700.
If the chemical elutes somewhere between
hexane and heptane then, by definition, the GCRI is somewhere between 600 and
700. Kovats proposed a formula for calculating the GCRI. Suppose the lower
n-alkane elutes at time A, the higher (n+1)-alkane elutes at time B and the
chemical elutes at a time X between A and B then:
Equ. 1‑6 GCRI = 100 * (
log(X)-log(A))/(log(B)-log(A)) + n * 100
If, in the example above, hexane eluted at
60 seconds, heptane at 80 seconds and the chemical at 70 seconds then GCRI =
100 * (1.845-1.778)/( 1.903 - 1.778) + 600 = 654
If we can predict GCRI from the chemical
structure then we can provide an accurate estimate of the retention time in a
specific column provided we know the retention times of the alkane series.
The simplest way to estimate GCRI is to say
it depends only on the boiling point of the chemical. This turns out to be an
impressively good predictor. The reason for this is simple – at the very
low concentrations of the chemicals in the GC the behaviour of the gases is
close to “ideal” so molecules hop on and off the support more or less according
to their volatility.
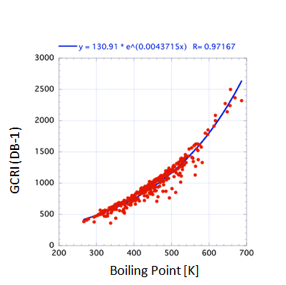
But “impressively good” isn’t “good
enough”. In the figure for GCRI values on a DB1 (Polysiloxane) support the
general trend with boiling point is clear, but when you look more closely at
the graph you see that for a given boiling point the GCRI can vary by 200-300
– much too large an error to be a useful predictive tool.
Clearly the assumption that the chemical is
behaving in an “ideal” manner is not working. And of course we know that
different chemicals will interact more or less strongly with the support phase.
And a good measure of that interaction is the HSP distance.
It then seemed obvious to add a correction
term that depended on the classic HSP distance calculation involving δD, δP and δH and
the values for the chemical, c, and support s. When this was tried, the results
were worse than using just the boiling point! What had gone wrong? One hypothesis
is that the δD value is already “included” in the boiling point (for example,
Hildebrand and Scott have shown that for simple alkanes, δD ²=RT+17.0*Tb
+ 0.009*Tb2). If the δD term
is removed from the distance calculation so that:
Equ. 1‑7 Distance² = (δPc-δPs) ² + (δHc-δHs)
²
then the fit becomes excellent:
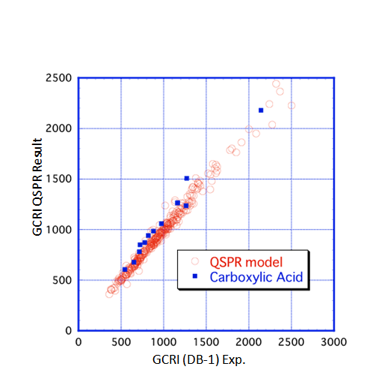
The correlation formula used (via a
Quantitative Structure Property Relationship model) is:
Equ. 1‑8 GCRI =
-220.27+209.51*(BPt*0.000519+1)^8.52* (DistPH*-0.0372+1)^0.156
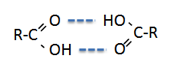
Well, it’s almost excellent. The main
exceptions are the carboxylic acids, shown
in blue. Perhaps this is because the acids tend to form dimers:
We’d like to claim that we have a universal
formula for GCRI. However, the only other extensive dataset available to us,
using the DX1 column, has a slightly different fitting formula which also
requires the MVol. We would need more datasets before being able to resolve
this dilemma. So the GCRI modeller lets you choose between these two (quite
similar) columns. If you try other values for the HSP of the columns we can’t
say what your predicted results
will be like. If you have access to other GCRI datasets for other columns, we’d
be delighted to fit them and try to get a more generalised rule.
In the HSPiP software we implement the
correlation via the Y-MB functional group method using neural network fits for
the boiling point and the HSP, though manual entry of the values is also
possible if they are known independently.
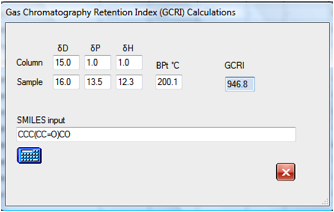
In this example the GCRI for a branched
aldehyde/alcohol is calculated as 947. If you wanted to test the purity of this
compound in a GC column you would simply set up conditions that gave you
reasonable elution times for nonane and decane and you would find your compound
nicely resolved. If you thought that your sample would have impurities with one
less and one more carbon in the main chain then the GCRI values for these two
molecules are quickly calculated as 845 and 1034 respectively so you would set
up your GC column for conditions where octane to decane (or undecane) eluted in
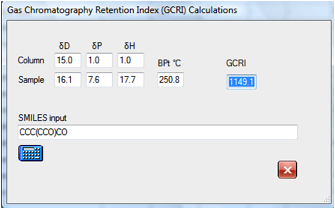
reasonable times. If you found a peak at around 1149 some simple
experimentation with likely contaminants show that it is likely to be the
dialcohol:
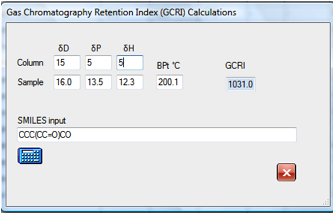
If you wanted to explore the effect of
changing to a support with different HSP values then it is simple to find that,
for example, that the original molecule will move from 947 to 1031 for a
slightly more polar column. Changes in the δD value make, of
course, no difference to the calculated GCRI.
HSPiP, therefore, provides you with a very
powerful predictive tool for your GC work.
HPLC
There are good theoretical reasons for
believing that High Performance Liquid Chromatography (HPLC) should conform exactly
to HSP for systems other than those that depend on ionized (acid/base) or
size-related separation. The principle is simple: the analyte has to “choose”
between residing in the stationary phase of the column and the mobile phase of
the eluent. And the “choice” is a simple HSP calculation. The parameter of
interest is k’, or “capacity factor” (which is calculated from the retention
time and the column volume, so a large k’ represents a large relative retention
time), and the key formula is:
Equ. 1‑9 ln k’ = C1 + C2 * MVol * (Distancema
– Distancesa)
where C1 and C2 are constants, m=mobile
phase, s=stationary phase and a=analyte, MVol is the molar volume of the
analyte and the distances are the classic square distances:
Equ. 1‑10 Distance2ma = 4*(δDm-δDa)2 + (δPm-δPa)2
+ (δHm-δHa)2
Equ. 1‑11 Distance2sa = 4*(δDs-δDa)2 + (δPs-δPa)2 + (δHs-δHa)2
What is surprising is that these simple
formulae have been used very little. Why is this? Sadly, the pioneering papers
were written on the basis of rudimentary Hildebrand solubility parameters. As
we now know, without the partitioning into D, P and H, the resulting
correlations simply aren’t reliable enough and the technique of correlating
with solubility parameter quickly fell into disrepute. Later attempts failed
because reliable values of HSP were hard to find and relatively small errors in
HSP lead to large errors in the predicted k’. For example, Ethyl benzene and
Methyl benzene (Toluene) are very close in HSP space and if we were interested
in them as solvents we would not be too bothered if a parameter were out by 1
unit. But for HPLC, Ethyl benzene and Toluene are classic test analytes which
separate readily and although the MVol surely has the most significant effect
(123/107), a small error in the HSP can rapidly be amplified by the squared effect
in the two distance calculations.
An excellent data set covering a good range
of analytes, columns and solvent mixes comes from Professor Szepesy’s group at
the Technical University of Budapest. The group have explicitly rejected
solubility parameters because they were not of sufficient predictive value.
It was a good test to use the most recent
HSP data set to see if it could do a good job in fitting the data. This is a
tough challenge as there is a lot of high-quality retention data to be fitted
using pure thermodynamics with no adjustable parameters. At the start of the
fitting process the key unknowns are the HSP of the column materials and the
HSP of the eluent. The prime data set uses 30/70 acetonitrile/water and it was
straightforward to confirm that by using the 30/70 mixture of the standard
acetonitrile and water values there was an adequate fit. The data could then be
processed to find that [15, 0, 1] provided an adequate fit for the first
stationary phase tried – reasonable for a classic C18 column. Other
columns in the data set surprisingly gave [15, 0, 1] as about the best fit,
including one –CN column that is supposed to be more polar. The
differences in k’ values between the columns depended strongly on the Slope
factor (C2). The variation from column to column made no obvious sense to us
till the correlation was found with the %C values (% Carbon) provided for each
column in the data set. Large %C’s gave large slopes (and therefore large
separations).
The challenge now got tougher given that
the fits had to work not only for one solvent blend but for a range. This was
initially unsuccessful. The changes in k’ values were larger than the change in
the HSP of the solvent blend would naturally produce. The literature provided various
suggested fixes for this much-observed phenomenon but nothing simple and
elegant suggested itself. The other problem was that the basic theory said that
the Slope value should be a constant, equal to 1/RT whereas it depended on the
column and was a factor ~25 smaller than 1/RT.
In the end a simple solution emerged that
gave adequate data fits across 60 data sets: 5 columns, two different solvents
(acetonitrile, methanol) and 6 solvent ranges (20, 30, 40, 50, 60, 70%
solvent). The Slope term was modified to C2 * (1-0.013 * % Solvent). So far there
is no theoretical explanation for this fudge factor nor for the fact that C2 is
out by a factor of ~25, but the fit to a large number of totally independent
data sets seems to be compelling evidence that the approach has many merits.
Having now fitted many 10’s of data sets
with a considerable degree of success, we need to point out that HPLC systems
do not operate in the sort of HSP zone to which we are all accustomed. The
reason for this is explained in a groundbreaking paper on using solubility
parameters (alas, ahead of its time) by Schoenmakers’ team in Chromatographia 15, 205-214, 1982. They
show that the stationary and mobile phases are very far apart in HSP space.
This amplifies the separation and (therefore) selectivity. What follows from
this is that the HSP of the stationary phase have a surprisingly small effect
on the separation. Changes in the mobile phase have a significant effect, but
even this is modest when you consider the vast change in HSP between, say, 100%
acetonitrile and 100% water. The clever thing about HPLC is that the only thing
which makes a big difference to retention time is the molar volume and HSP of
the analyte. It’s worth repeating that the modest effect of mobile and stationary
phase and large effect of HSP falls very naturally from Schoenmakers’ analysis.
What’s not so obvious is the very
significant effect from molar volume. The large separation between toluene and
ethyl benzene or between the various parabens also included in the Szepesy data
set comes almost entirely from the molar volume effect. One obvious explanation
for this is some sort of diffusion limited or size-exclusion effect. But that’s
not what the basic theory says. The molar volume term comes in simply because
the thermodynamics say that interaction energies are HSP/Molar_Volume. This
large effect and its scientific explanation are a particularly satisfying
vindication for the HSP approach.
Here are two plots that show the reasonable
degree of success of the technique. The data are for columns at either end of
the spectrum. The first is a classic hydrophobic C18 column and the second is a
classic polar CN column. The results are based on an optimum HSP of [15, 0, 1 ]
value for both stationary phases as discussed above. For the CN column the
retention times are rather small, particularly for the left-hand side of the
graph. In the original paper, the authors discarded the data points for
Caffeine and Hydroquinone because their retention times were too small. In each
case the plots are of actual v predicted ln k’. Below the plot of the fit is an
indication of what the idealised HPLC trace would look like with the fitted
values. The value of this will emerge in a moment:
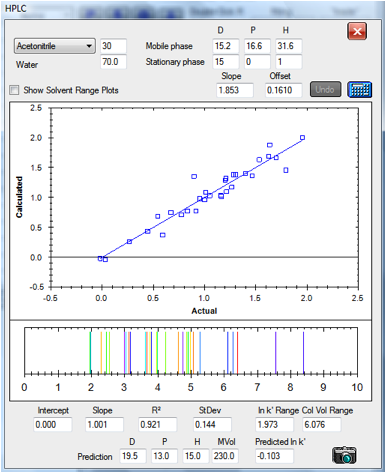
Figure 1‑4 A reasonable correlation for
the C18 column:[15, 0, 1]
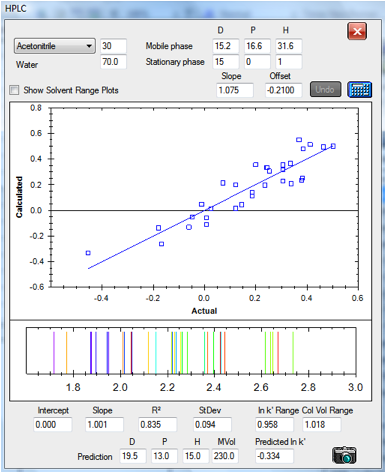
Figure 1‑5 The correlation for the CN
column: [15, 0, 1] It’s noisier partly because it spans only a small range of
column volumes. The data set is slightly different from the one above,
reflecting slightly different experimental protocols in the original papers
The software allows you to interact freely
with the data. You can create mobile phase parameters directly by choosing the
solvent (Acetonitrile, Methanol or THF) and the relative percent with water
– or you can directly enter your own values. The Calculator button
automatically finds a slope/offset combination that give you a slope of 1 for
the best fit line. You can alter these values manually if you wish.
The output includes the plot itself (moving
the mouse above a data point reveals the solvent, actual and calculated data)
then the intercept, slope, R² (goodness of fit) Standard Deviation and
then the range over which the separation works in terms of k’ and in terms of
column volumes. You can see that the –CN column fits all the chemicals in
a range of 1-2.018 column volumes whereas the C18 covers 1-7.076 column volumes
– giving you a choice of speed versus resolution.
The same C18 column run with 60% acetonitrile
gives a relatively good fit. No fitting parameters were changed compared to the
30% values:
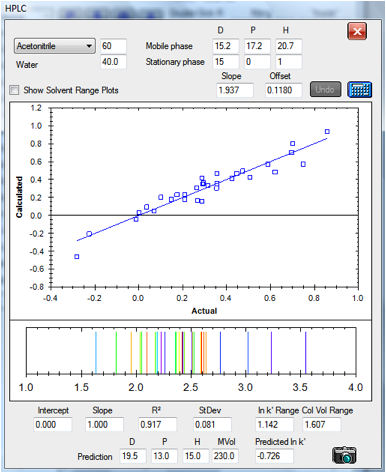
Figure 1‑6 The correlation for the C18
column at 60/40 acetonitrile/water
The fact that the same fitting parameters work
for a large shift in the solvent blend indicates the power of the technique.
You can play live with these data yourself
(plus the other HPLC data we have provided) and reach your own conclusion. One
thing that playing “live” means is that you can alter the individual HSP from
the standard HSPiP main form and see the effect on the HSP fit. We did this
ourselves. The first was to fit p-Nitrophenol. It was obvious on the plot that
the fit was very poor and we played with the values till the fit seemed
reasonable. Only then did we realise that the published value for p-Nitrophenol
was, regrettably wrong; the δP of 20.9 simply makes no sense. The second was to
play with Caffeine - the point nearest the 1 column volume elution point and
therefore the least accurate. The official value is an estimate. Perhaps the
HPLC data are telling us to revise that estimate. The final adjustment was to
Pyridine. The published value was re-checked and makes good sense, but was way
off in the plots. When we looked at other examples of Pyridine in HPLC we noted
that it was normally eluted as Pyridinium as the mobile phase is typically
buffered to pH 3.0. Although the Szepesy data set was explicitly run
unbuffered, we suspect that the value we have entered is for Pyridinium rather
than Pyridine.
At this point the reader can go in two
directions. The first is to say “They’ve fiddled the data to get the answer
they wanted”. The response to that is that most “fiddling” with the data,
although it could improve any single plot, generally made the plots over the
full Szepesy data set positively worse. The second direction is to say “This
looks like a powerful technique for understanding and predicting HPLC behaviour”.
That’s why we’ve added the k’ range output (so users can think which column
and/or solvent blend to use) and also the predictor. You can see in the screen
shot how an analyte of [19.5, 13, 15] with a molar volume of 230 would behave
in each of these columns.
The second direction makes sense. HSP and
HPLC are a powerful combination – for predicting/understanding HPLC and
for generating HSP values for use in other applications.
That’s also why the simulated elution plot
has been included. When you change the % solvent (or the solvent itself) you
can see how the eluted peaks are predicted to change. You can quickly see how
two peaks close together can be separated or vice versa as you change the
elution conditions. Similarly, if you click the Show Solvent Ranges you get a
graphical idea of how the k’ of the individual analytes vary over the full
0-100% solvent range.
Expert HPLC users will object that the
predictions of k’ are unlikely to be precise enough – they are, after
all, predictions of ln k’, which can easily compress errors. This may well be
true. But look at what the HSP approach offers. With one rather simple formula,
with very few fitting parameters (and maybe after more investigation, no
fitting parameters at all), a user or manufacturer can understand numerous
fundamental aspects of what is going on in a separation. It seems, at the very
least, that the HSP-HPLC approach should be investigated more deeply with
alternative data sets.
Finally, here’s a data set prepared
independently for us by Andrew Torrens-Burton, an expert Analytical Chemist at
MacDermid Autotype. He gathered data for three different types of analyte,
deliberately avoiding the aromatic test analytes used in the Szepesy data. The
standard values for the C18 column calculated from the Szepesy data set were
used for this analysis except that the slope is bigger – because this
column happened to be twice the length of that used by Szepesy:
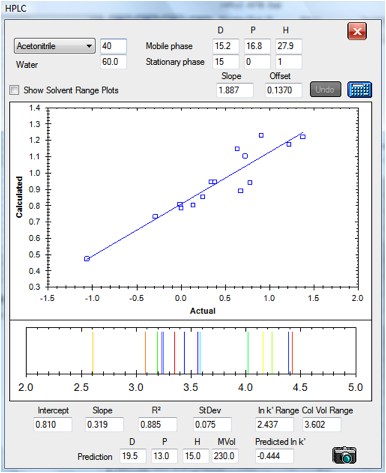
Figure 1‑7 The correlation for the set
by Andrew Torrens-Burton
NMR
Solvents
This is a very short section. We have not,
so far, attempted any correlations between NMR and HSP, though it is clear
that, for example, polymer-polymer interactions in solution will depend
strongly on polymer-solvent interactions which can, in turn, be predicted via
HSP.
But we see a very powerful use of simple
HSP ideas to help the practical NMR spectroscopist.
One typical challenge is to find a solvent
for carrying out NMR experiments for a given chemical. There are relatively few
H-free solvents (i.e. deuterated chloroform, benzene etc. plus carbon
tetrachloride) and if your chemical is not very soluble in any of them then you
have problems.
But we know from HSP theory that a chemical
can be insoluble in each of two solvents yet be soluble in a mixture, if that
mixture is a good match for the HSP of the chemical.
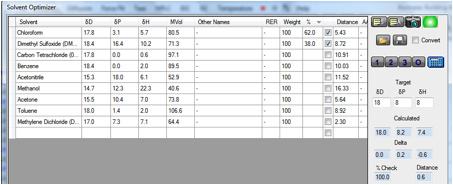
We’ve therefore provided a Solvent
Optimizer dataset containing the most used H-free (deuterated) solvents.
If you enter the Target HSP of your
chemical (perhaps estimated via Y-MB) and click the 2 button you instantly find
a blend that gives you an optimum match. To be more precise you can click the 3
button to find a 3-solvent blend. If you are worried about cost you can even
weight the solvents (high cost = low weight) so that the optimum blend uses
less of the more expensive ones.
Armed with those data it is highly likely
that you will find it much easier to get a higher concentration of your sample –
at very little cost and very little trouble. In the following example a tricky
[18 8 8] material is likely to be nicely soluble in the chloroform/DMSO mix
– and it is highly unlikely that you would have tried that mix without
help from the Solvent Optimizer.
E-Book contents | HSP User's Forum
