Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
In earlier editions of HSPiP we took it for
granted that the explanation in the previous chapter was sufficient to help
users calculate their first solubility Sphere. But we found that users were
very unsure of themselves and we often had to email advice to them. Thinking
back, one of us (Abbott) remembers how nervous he was with his first Sphere, so
the following practical guide reflects his memories of his first Sphere, backed
up by the experience of measuring many different Spheres over 40 years by
Hansen.
Let’s start with a typical example. You
want to dissolve a polymer for some sort of processing such as coating. You
have severe restrictions on cost, health & safety or environmental impact
so finding a good solvent blend is very hard. If, instead, you are interested
in dispersing a pigment or nanoparticle, the discussion below is identical,
just substitute “pigment” or “nanoparticle” for “polymer”.
Our task is to define the HSP of the
polymer. A solvent (blend) that is close to that HSP will be a good solvent.
Here’s how to do it.
Get about 20 glass vials. Put a small
sample of the polymer into each of the vials. Now add a small amount (say 5ml)
of a different solvent to each of the vials. Which solvents should you use?
Well, the following list is a pretty good mixture of relatively common and
relatively safe solvents. It’s included as Test Solvents.hsd with the package
and you can easily add/remove solvents to suit your needs.
|
Solvent |
D |
P |
H |
|
1.4-DIOXANE |
19 |
1.8 |
7.4 |
|
1-BUTANOL |
16 |
5.7 |
15.8 |
|
2-PHENOXY
ETHANOL |
17.8 |
5.7 |
14.3 |
|
ACETONE |
15.5 |
10.4 |
7 |
|
ACETONITRILE |
15.3 |
18 |
6.1 |
|
CHLOROFORM |
17.8 |
3.1 |
5.7 |
|
CYCLOHEXANE |
16.8 |
0 |
0.2 |
|
CYCLOHEXANOL |
17.4 |
4.1 |
13.5 |
|
DBE |
16.2 |
4.7 |
8.4 |
|
DIACETONE
ALCOHOL |
15.8 |
8.2 |
10.8 |
|
DIETHYLENE
GLYCOL |
16.6 |
12 |
20.7 |
|
DIMETHYL
FORMAMIDE |
17.4 |
13.7 |
11.3 |
|
DIMETHYL
SULFOXIDE |
18.4 |
16.4 |
10.2 |
|
DIPROPYLENE
GLYCOL |
16.5 |
10.6 |
17.7 |
|
ETHANOL
99.9% |
15.8 |
8.8 |
19.4 |
|
ETHYL
ACETATE |
15.8 |
5.3 |
7.2 |
|
GAMMA
BUTYROLACTONE |
19 |
16.6 |
7.4 |
|
HEXANE |
14.9 |
0 |
0 |
|
MEK |
16 |
9 |
5.1 |
|
METHANOL |
15.1 |
12.3 |
22.3 |
|
METHYL
ISOBUTYL KETONE |
15.3 |
6.1 |
4.1 |
|
METHYLENE
DICHLORIDE |
18.2 |
6.3 |
6.1 |
|
n-BUTYL
ACETATE |
15.8 |
3.7 |
6.3 |
|
N-METHYL
PYRROLIDONE |
18 |
12.3 |
7.2 |
|
PM |
15.6 |
6.3 |
11.6 |
|
PMA |
15.6 |
5.6 |
9.8 |
|
PROPYLENE
CARBONATE |
20 |
18 |
4.1 |
|
TCE
TETRACHLOROETHYLENE |
18 |
5 |
0 |
|
TETRAHYDROFURAN |
16.8 |
5.7 |
8 |
|
TOLUENE |
18 |
1.4 |
2 |
But if you don’t have all of them, no
matter. And if you have some others that’s fine. Just don’t have too many of
the same thing. It probably doesn’t help to have methanol, ethanol, propanol
and butanol or pentane, hexane, heptanes and octane. Just ethanol and hexane
will be good enough.
Now you need to find out in which solvents
the polymer is soluble. Here you have to make a decision. You can, for example,
just hand shake each sample and then see which ones give clear solutions and
which ones don’t. But often you find that such a test is useless as even good
solvents might take some time to dissolve your polymer. So you might decide to
put all the vials into an ultrasonic bath for 10 minutes then inspect each vial
as soon as it comes out of the bath. But often you find that everything gets
dispersed by the ultrasonics so that everything looks to be “soluble”. So you
might decide to check after the
samples have sat for 10 minutes at room temperature. And maybe the polymer
doesn’t “dissolve” in any of the solvents (perhaps you had too much polymer or
too little solvent) but you can still see that the polymer is highly swollen in
some solvents and completely unswollen in others so you can use that as your
criterion for “good” and “bad” solvents. For some polymers you might even have
to wait for days to distinguish “good” and “bad”.
Don’t despair if the previous paragraph is
too vague for you. You are a scientist and you probably already have a good
intuition about the general solubility properties of your polymer, so you can
make a good decision about what treatment to adopt and whether you judge by
dissolution or by swelling.
One word of advice. Because the effects of
temperature on solubility can be quite complex, stick with room temperature
tests if possible – at least till you have gained some experience in the
whole process.
And if you aren’t interested in polymers
but in, say, pigments, the above description applies to you too. Just find a
set of conditions where your pigment is obviously “happy” in some solvents
(e.g. nicely and permanently dispersed) and “unhappy” (e.g. sitting as a lump at
the bottom of the vial) in others. Be careful to check if any pigment is stuck on
the side of glass containers.
If you are lucky (or already have a good
intuition) after a moderate effort you now have a list of good and bad
solvents. If your system is entirely new then you might spend a day finding the
appropriate test conditions, sometimes getting sidetracked when a “clear”
solution is in reality a blob of polymer stuck under the cap of the vial. But
once you have your list of good and bad solvents then the Sphere calculation
does the rest.
With all the scores entered, 1 for good and
0 for bad, you click the Calculate button and you get two vital bits of data,
both of which are important. The first is the HSP of the polymer. That’s
defined as the centre of the Sphere. Any solvent close to that centre will be
excellent for that polymer. But how close is “close”? That’s why you need the
second bit of data which is the radius of the Sphere. You’ll remember that RED
number is the ratio of the distance of a solvent from the centre of the Sphere,
divided by the radius of the Sphere. If your polymer gives a small radius, say,
4, then a solvent with a distance of 4 is just on the boundary – the RED
is 4/4=1. A solvent with a distance of 8 is therefore a bad solvent, with a RED
of 8/4=2. But if your polymer is more forgiving, then the radius might well be
8. So the solvent with distance 4 now has a RED of 4/8=0.5 which means that
it’s likely to be pretty good. The solvent with the distance of 8 now has a RED
of 8/8=1 so is borderline.
The previous paragraph is so important that you need to read it again
till you’re 100% clear. Both the centre and the radius of the Sphere are
vital for you to know. Later on, when you understand the Polymers form in HSPiP
you’ll find some tricks for working out which solvents (usually from a list
that is different from the one you used to determine the Sphere) are good or
bad. By adding your polymer (both HSP and
Radius) to the Polymers form with one click you’ll be able to sort your solvent
list in order of RED – with the low REDs being the good solvents.
Here’s an example (included in HSPiP) where
the Sphere comes out best matched to chloroform. You can instantly tell that
it’s the best match because the solvents are sorted by their RED numbers and
chloroform has the smallest RED. In later chapters we will see how you can find
a solvent blend that would be a close match to chloroform and the centre of the
Sphere, but without the (probably) unacceptable use of chlorinated solvents.
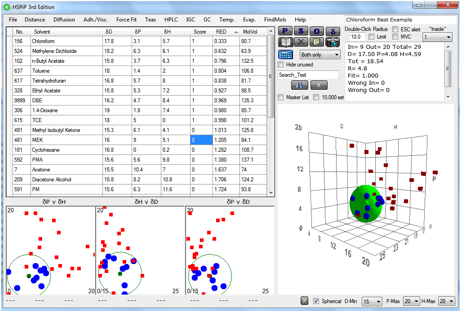
Figure 0‑1 A typical first Sphere test using a typical, sensible range of
solvents
Bad
Spheres
There are two types of bad Spheres that
come out of such experiments.
1 The first is a Sphere with (approximately)
the same values each time you click the Calculate button, but with an
appallingly bad fit – with many good solvents outside and bad solvents
inside. In general there are three possible reasons for this
·
You’ve made some
misinterpretations of your data. It’s quite common, for example, to go back to
recheck a “good” result which doesn’t fit the Sphere, only to find that the
polymer is stuck underneath the lid of the test-tube rather than, as we
thought, being nicely dissolved. When you correctly class this as “bad” the
Sphere fit greatly improves
·
HSP just don’t apply to your
system. This is possible, but unusual. Pigments with highly charged surfaces
may fall into this category
·
Your test material is impure.
In fact we have often proved that
materials are impure by finding bad HSP Spheres and alerting researchers to
look out for (and find) impurities
·
Your material has a split
personality – e.g. a 1:1 block copolymer made from hydrophilic and
hydrophobic parts. In principle we could force HSPiP to generate two Spheres,
but we’ve never been convinced of the value of this. But see v3.1!
2 The second problem shows up as a Sphere
which appears in a different place each time you click the Calculate button.
This is simply a case where you don’t have enough good/bad solvents surrounding
the centre of the Sphere to create an unambiguous best fit. For example, if the
real δD is ≥19, the chances are that you’ve used very few solvents up in
this range. There is no way to know if the real δD is 19 or 20 or 21 because
there are no δD=21 solvents giving you a “good” or “bad” result. The only way
you can pin down the real Sphere is to work out (it should be obvious looking
at the 3D plot and the 3 2D plots) where you are lacking data. Then simply find a few relevant solvents
and do the tests. We mentioned earlier that sometimes you have to use nasty
solvents in tests. You may well be forced to use such solvents to get data from
a relevant region. As long as you can handle the solvent safely for the test,
it doesn’t matter that it too nasty to be used in a real application. Remember
that you don’t have to re-do any of your previous tests. You simply add the new
data to your list and click the Calculate button.
This still doesn’t explain why the “best
fit” moves around so alarmingly. This is for two reasons – one
mathematical, one philosophical
HSP 3D space is quite complex. If you could
test each point in that space to see if it’s the best (or least bad) fit to the
data you would find in the case of really good data sets that there is a clear,
deep well down into which any good fitting algorithm would quickly fall,
finding the same result each time it started, from wherever it started. With
under-specified datasets, there are no such deep wells and it’s very easy to fall
into a shallow well, thinking that it’s the best. So the “best” fit depends on
your starting assumptions. This is typical mathematical behaviour.
Philosophically we could make ourselves
look good by always leading you to the same false fit, however often you
clicked the Calculate button. But we deliberately want you to see when the fit
is poor. The fact that you see a different “best” fit each time is telling you
clearly that the dataset is under-specified and that you need to gather more
data points if you really want to know the centre and radius of the Sphere.
Each time you click the Calculate button, the fitting algorithm starts from a
totally different part of HSP space and therefore, in the case of
under-specified data, is likely to end up in a different “best” fit each time.
However, for the 3rd Edition
we’ve been able to find a better way to search the whole 3D space and the
“jumping around” problem has been much reduced.
Changing
the fitting algorithm
The “classic” Hansen fitting algorithm has
been used successfully for over 40 years. It systematically explores the whole
of HSP space and weighs the errors of good-out and bad-in depending on how
badly out(in) a solvent actually is. This makes a lot of sense. But one very
wrong point can exert a disproportionate effect over the fit. We’ve therefore
provided the option of a totally different way of identifying and finding a
best fit. The GA (Genetic Algorithm) attempts to find the least wrong solvents
within the smallest possible radius. The search is via the genetic approach
using “genes” that create the fit and selecting better genes throughout the
generations. Not surprisingly, the two approaches often reach a near-identical
result and the classic approach tends to be faster than the GA algorithm in these
cases. But in other situations (e.g. with odd outliers) the GA result seems
more intuitively satisfactory than the classic approach. You, as a scientist,
can reach your own judgement for your own cases.
Changing
the definition of “good”
A lot of people are worried about the
flexible definition of “good”. It doesn’t sound precise enough to be good
science. So as an exercise, take the above sample file and deliberately set two
of the solvents near the edge of the Sphere (RED ≥0.98) to “bad”. This could
happen, for example, if a colleague looked at your test tubes and said “I think
you are being too generous in your evaluations. I don’t think that dioxane and
TCE are good – they should be classed as bad”. When you calculate the
Sphere you get new values [17.10, 3.07, 5.10, 4.10].
The changes in this case are typical of
what you find in real-world examples. The centre of the Sphere does not change
all that much, but the radius changes, in this case by quite a lot (from ~8 to
~4). This is an important result. The key “truth” about this polymer reside in
the HSP. These don’t change much when you change your definition of borderline
good/bad. The radius is an important part of the HSP characterisation of a
polymer, but it must be variable
– for example a lower molecular weight version of the same polymer will
be more soluble and have a larger radius.
Please do try this playing around with the
data. If you eliminate more and more solvents then, of course, the Sphere will
start to move around more. You can’t be too careless about the definition of
good. And when you are down to just a few good solvents, the data become
statistically less satisfactory. If you find yourself in this situation you
have no other choice than to find a few more solvents in the critical region
and testing those for good/bad. If you find one or two more good solvents then
your confidence in the HSP of the polymer will improve.
Rational
improvement
One user of HSPiP asked if we could add a
feature to help rationally improve the quality of the fit. What he wanted was an
automatic scan of the 3D fit to identify “diversity voids”, i.e. areas in 3D
space where there is no relevant data. For HSP, “relevant” means ‘close to the
edge of the Sphere and in an area where there are no other solvents’. Why is
this important? If you test extra solvents that are either near the centre of
the Sphere or near another solvent or are far outside the Sphere, you get very
little extra information. But if you find a solvent near the edge, and in a
direction where there are no other solvents then this one extra data point
could be crucial for defining the edge.
We’ve found a relatively simple way to
implement this process. A file called SphereCheckMaster.hsd contains a list of
80+ solvents that fill a good part of 3D space. These solvents are a subset of
the original Hansen 88 solvents so their values are well-validated. When the
Sphere fit has taken place, the software scans each of the solvents to see if
it is near the edge of the Sphere (i.e. 0.9<RED<1.1) and if it is whether
it is far enough away from the other solvents used for the fitting. If both
those tests are passed then the solvent is shown in the plots in orange and
added to the table of solvents. As an intelligent user you can then make the
judgement as to whether any or all of those solvents would really be worth
testing. If you are happy with “good enough” then you may not even want to do
this Sphere Radius Check (SRC). If you do the check you might disagree with the
program’s judgement on some of the solvents but might agree that one or two of
the solvents might be worth doing. You might then find that one of these
solvents is unacceptable in your lab. At that point you can highlight that
solvent and go into Solvent Optimizer to find if there is a blend of nicer
solvents that could be used instead.
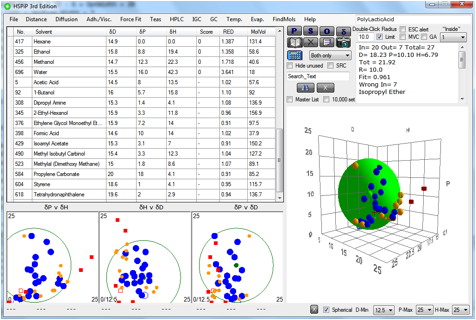
Figure 0‑2 12 solvents (in orange) suggested by the SRC check.
In summary, the SRC is a tool that you are
free to use to help you decide on whether it’s worth doing more experiments.
It’s certainly not a perfect tool (there are probably more sophisticated
algorithms that could be used) but we find it helpful so we hope that you will
too.
Using
numeric data for greater accuracy
If you can go to the trouble of measuring
actual solubility of the polymer rather than Yes/No solubility then you have
richer data and the chance to get a more accurate fit. The Data fit method in
HSPiP will, therefore, provide a more accurate value, especially if fewer
solvents are used. Clearly there is a trade-off: there is more work required to
measure the solubilities, but less solvents (if chosen carefully) are required.
An alternative numeric approach uses
measurements of the intrinsic viscosities of the polymer in the solvents. The
idea behind this is that the better the solvent, the higher the viscosity
(because the polymer opens out more). The equations for fitting to intrinsic
viscosity data are simple as discussed in, for example, Farhad Gharagheizi,
Mahmood Torabi Angajia, A New Improved
Method for Estimating Hansen Solubility Parameters of Polymers, Journal of
Macromolecular Science, Part B: Physics, 45:285–290, 2006. If HSPiP users
would like us to implement those equations we would be happy to do so.
Remember, however, that the use of limited
numbers of solvents comes with the danger of not encompassing sufficient of HSP
space. Fitting algorithms can always find an optimum, but if there are no
solubility data in important parts of HSP space then these optima will be
false. Many polymers have one or more of the HSP higher than any potential test
solvent. Simple averaging the HSP of the test solvents, weighted in any way,
cannot lead to the correct answer. The sphere fitting process is required. The
intrinsic viscosity inherently limits a study to those solvents that do
dissolve the solute. There are in essence no “bad” solvents for measurement of
the intrinsic viscosity, but ranking the intrinsic viscosity into the standard
1-6 ratings, possibly with a 0 for those solvents known not to dissolve the
solute, will allow an analysis in the usual way.
E-Book contents | HSP User's Forum
