Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
Chapter 31, Predictions (Many Physical Properties are Correlated with HSP)
HSE
The HSE modeller captures in one place all
the capabilities we have for making rational choices about chemicals in terms
of Health, Safety and Environment. It lets you enter the SMILES for two
chemicals and then lets you compare a large set of important properties. A
typical example is “read across” for REACH and other chemical safety systems.
If you know that a particular chemical is safe or unsafe then a rational
starting point for judging the safety properties of an un-tested material is to
“read across” from this chemical to a similar one. But how similar is similar?
Only you as a scientist can judge, but in the HSE comparison you can compare
estimates of
·
Phase change properties –
melting point, boiling point, vapour pressure
·
Solubility properties –
solubility in water, Octanol/Water partition coefficient, Soil/Water partition
coefficient, BCF (Bio Concentration Factor) for Fish oil
·
VOC properties – RER,
vapour pressure, flash point, OH radical reactivity, Carter MIR
·
Other properties – Heavy
Atom Count, Density, Molecular Weight, Molar Volume
·
Numerical comparison –
HSP distance, “Functional Group distance”
The functional group distance is an
estimate based on the (dis)similarity of the functional groups derived from the
Y-MB analysis. If, for example, the two molecules both of FG#27 (primary
alcohol) then their distance is lower than if one has FG#27 and the other has
FG#38 (primary amine) which in turn is larger than between FG#27 and FG#28
(secondary alcohol). The methodology takes into account the different molecular
sizes and the numbers of functional groups. Clearly a molecule with many
functional groups must be quite distant from one with just a few, even if those
few match groups in the larger molecule. To ensure that differences aren’t too
exaggerated, although butanol has 4 carbons and methanol has only 1, each molecule
is shown as having just two functional groups, and the distance between methyl
and butyl is not all that large. This calculation is different from one which
would count similar methyl and alcohol groups in both molecules but would have
two un-matched CH2 groups in the butanol.
A
note on LogP=LogKow=Octanol/Water partition coefficient
LogP is often seen as a highly important
parameter. Although it is important we think that it is very much misused. In
our view, HSP are very often much more insightful than LogP. The main reasons
we are sceptical about LogP are:
·
It is a ratio which can hide
important details. A LogP of 0 (i.e. P=1) could have solubilities of 100/100 or
0.001/0.001. Chemically the former (high solubility) is likely to be very
different from the latter (low solubility) even though the ratio is the same.
·
Chemicals in biological
environments don’t have a choice between a “water” environment and an “octanol”
environment. A typical lipid environment might be much closer to [16, 3, 3]
than octanol’s [16, 5, 12] and as we showed in another chapter, a lot of key
biological entities such as skin or DNA binding sites are closer to [17, 10,
10] than to octanol. LogP is far too restricted to be able to give a reliable
guide to where a chemical might be going.
But as a service to HSPiP users we felt it
was important to provide the best-possible predictor. Hiroshi’s www.pirika.com has a long article on his
search for the best predictor of LogP. Not surprisingly (see the section on the
hydrophobic effect on solubility below) the best single predictor for LogP is
MVol. If you plot the LogP values of many different classes of molecules (e.g.
hydrocarbons, nitro, amide, nitrile, amine ) you get a series of straight
lines. So LogP has the same linear dependence on MVol, though with a different
offset for each functional group:
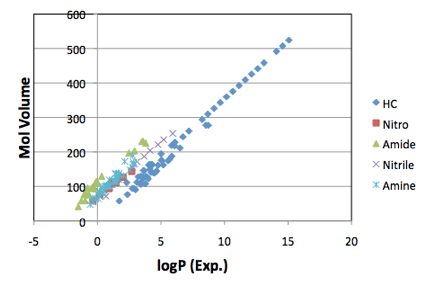
Figure 1‑1 A typical example of linear correlations between MVol and logP. The
slopes are the same, with different offsets. The original article at www.pirika.com has many more examples.
Armed with this knowledge it is possible to
do a more exact prediction of LogP taking into account the offsets from the
different functional groups. This requires the offsets to be additive which,
fortunately, they happen to be. With the functional group correction, the
correlation between LogP and MVol is very strong:
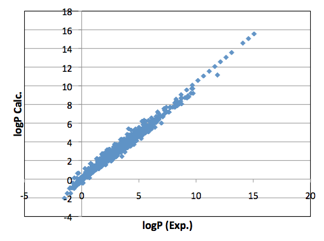
Figure 1‑2 The full correlation of 5,320 experimental LogP values against MVol
with functional group correction using the Y-MB functional groups.
Within the typical range of 0-5, the
predictions span a range of +/- 1 LogP unit. Given the inevitable uncertainties
in the experimental values this is an impressive fit.
Azeotropes
and Vapour pressures
If a mixture of two solvents were “ideal”
then the partial vapour pressures above the mixture would simply depend on the
saturated vapour pressure and mole fraction of each solvent.
But we know that in most cases the presence
of one solvent tends to make the other solvent “uncomfortable” creating a
higher-than-expected vapour pressure. The difference between ideal and real is
the Activity Coefficient. So to know everything about the partial vapour
pressures of a mixture, the activity coefficients have to be known.
No perfect way has been found to predict
activity coefficients, γ. Simple theory suggests that they can be calculated
directly from HSP using the formula
Equ. 1‑1 ln(γ)= ln(φ1/x1) + 1 - φ1/x1
+ χ12* φ22
where φ1 and x1 are
the volume fraction and molar fraction of solute 1, φ2 is the volume
fraction of solvent 2 and the parameter, χ12 is given by:
Equ. 1‑2 χ12 = MVol /RT * ((δD2-δD2)2
+0.25* (δP2-δP1)2 +0.25*(δH2-δH1)2)
In the absence of any better formula, this
is a good-enough approximation, but checking against a large dataset of
activity coefficients shows that it needs considerable improvement.
At the heart of the problem is that fact
that the basic formula does not account for positive interactions between
solvents that create activity coefficients less than 1. And a detailed analysis
of the failures from predictions of the simple formula show that the biggest
deviations typically come about amongst solvents with large δP and δH
parameters.
One way to fix this problem is shown by
MOSCED – Modified Separation of Cohesive Energy Density. This splits δH into
donor/acceptor terms. This seems a good idea. Unfortunately, MOSCED has not
become a generally acceptable methodology and some of the more recent fittings
of the complex parameters mean that the sum of the cohesive energy terms are
often very different from the cohesive energy – in other words, MOSCED
has become more of a fitted parameter technique than one rooted in
thermodynamics.
In the absence of any breakthrough theory
we’ve done a NN fit to a large database of Margules parameters. We have found that
the Margules formulation of activity coefficients is more useful than relying
on “infinite dilution activity coefficients”, especially as we are often
interesting in large mole fraction solubilities.
For Solute 1 in Solvent 2, the activity
coefficient γ1 for mole fraction x1 (and therefore x2=1-x1)is
given by:
Equ. 1‑3 Ln γ1=x22(Marg12+x1(Marg21-Marg12))
And of course it’s the other way round
for Solute 2 in Solvent 1:
Equ. 1‑4 Ln γ2=x12(Marg21+x2(Marg12-Marg21))
Armed with better predictions across the
whole mole fraction range it then becomes simple to calculate the isothermal
vapour pressure curves and only slightly more complex to calculate the vapour pressures
at the (variable) boiling point of the mixtures from which it is possible to
identify important azeotropes.
Solubility
It seems odd to say that you cannot
directly predict solubility from HSP! But HSP have always been about relative
solubility and have never attempted to issue exact solubility predictions.
However, with some simple equations and
some good estimations of key properties, it is possible to predict solubilities
directly.
The equation is simple:
Equ. 1‑5 ln(Solubility)= -C + E –A - H
C is the “Crystalline” term, sometimes
(confusingly) called the Ideal Solubility. It is the Van ‘t Hoff (or Prausnitz)
formula that depends on the difference between the current temperature, T, and
the melting point Tm, the Gas Constant R and also on the Enthalpy of
Fusion ΔF.
Equ. 1‑6 C = ΔF/R*(1/Tm – 1/T)
In other words, the higher the melting
point and the higher the enthalpy of fusion, the more difficult it is to
transform the solid into the dissolved (liquid) state.
This formula is a simplification which
follows convention and ignores some other terms like heat capacities. An even
simpler formula, from Yalkowsky, uses just the melting point:
Equ. 1‑7 C = -0.023*(Tm –T)
Recently, Yalkowsky has reviewed the
various options for calculating this term, S.H. Yalkowsky, Estimation of the Ideal Solubility (Crystal-Liquid Fugacity Ratio) of
Organic Compounds, J. Pharm. Sci, 2010, 99, 1100-1106 and confirms that
-0.023*(Tm-T) is good enough. The
paper uses Log10 so the printed coefficient is -0.01.
For calculations where Tm≤T,
C is zero.
The E term is (combinatorial) Entropy. This
is calculated from volume fractions (Phi) and molar volumes.
Equ. 1‑8 E = 0.5*PhiSolvent*(VSolute/VSolvent-1)
+ 0.5*ln(PhiSolute + PhiSolvent*VSolute/VSolvent)
It’s worth making an important reminder
that molar volumes for solids are not
based on their molecular weight and solid
density. In the words of Ruelle: “(For a solid) the molar volume to consider is
not that of the pure crystalline substance but the volume of the substance in
its hypothetical subcooled liquid state.”
A comes from the activity coefficient. The
larger the activity coefficient, the more negative A becomes. As discussed
above, a simple version of A can be calculated directly from HSP, but the more
sophisticated Margules formulation gives better predictions.
H is a Hydrophobic Effect term that is very
important for solubilities in water, and somewhat important for solubilities in
low alcohols. The calculation follows the method of Ruelle (see, for example, Paul
Ruelle, Ulrich W. Kesselring, The
Hydrophobic Effect. 2. Relative Importance of the Hydrophobic Effect on the
Solubility of Hydrophobes and Pharmaceuticals in H-Bonded Solvents, Journal
of Pharmaceutical Sciences, 87, 1998, 998-1014) and depends on rs*PhiSolvent*VSolute/VSolvent
with extra terms depending on how many hydrogen-bond donors (alcohols,
phenols, amines, amides, thiols) are on the solute and whether the solvent is
water, a mono-alcohol or a poly-ol. The value rs is 1 for
monoalcohols and 2 for water and, for example, ethylene glycol. It is 0 for all
other solvents. If the solvent is water and the solute contains alcohol groups,
there are special parameters depending on whether the alcohols are primary,
secondary or tertiary. There is a further refinement (not included in this version)
which discounts some of the solute’s hydrogen bond donors if they are likely to
be internally bonded. The important thing about the Ruelle formula is that
solubility in water depends almost entirely on the size of the solute –
bigger molecules are simply less soluble than smaller ones. Their explanation
is more sophisticated than the simple idea that bigger molecules disrupt more
hydrogen bonds, but the simple intuition isn’t a bad approximation. They show
that for “simple” molecules (one’s without too many –OH groups) spanning
a huge range of solubilities, a first principles formula based on MVol, with no
fitting parameters, does an excellent job at prediction.
The complication is that the E, A and H
terms all depend on the volume or molar fraction which is precisely what you
are trying to calculate, so there is an iterative process involved until the
equation balances.
Although the output of most interest is the
real solubility, it is very instructive to see the effect of the different
terms, so the HSPiP modeller shows the C, E, A and H terms. For all solvents
that aren’t water or alcohols H is zero. For water the H term, not
surprisingly, can be very large. But because of water’s small molar volume, the
E term can also be large. Because the A term can also be large, water
solubility is hard to judge a priori because it can involve the (partial)
cancellation of large numbers.
A very helpful way to think through
solubility issues has been provided P. Bennema, J. van Eupen, B.M.A. van der
Wolf, J.H. Los, H. Meekes, Solubility of
molecular crystals: Polymorphism in the light of solubility theory, International
Journal of Pharmaceutics 351 (2008) 74–91. The equations below can be
switched on and off in the Crystalline Solubility Theory modeller and plots can
be chosen as x v T (so both are in “normal” units) or as ln(x) v 1/T which is
the van’t Hoff plot which gives a straight line (added as a reference to the
plot) for ideal solubility, making it easier to see the effects of switching on
and off the different parameters. The Yalkowsky approximation is included for
reference.
For the ideal
solubility case the mole fraction solubility x is given by the equation we
have used earlier:
Equ. 1‑9 Ln(x) = ΔF/R*(1/Tm – 1/T)
However, this
assumes that the heat capacity Cp of the virtual liquid at temperature T is the
same as that of the solid. In general the heat capacity is higher so ΔCp is positive.
This happens to increase the solubility, sometimes to a surprisingly large
extent via:
Equ. 1‑10 Ln(x) = ΔF/R*(1/Tm – 1/T) + ΔCp/R [Tm/T – ln(Tm/T)-1]
If regular solution theory is used then there
is an additional term that depends on ΔHmix, the enthalpy of mixing and ΔSmix, the
enthalpy of mixing. If this is positive (i.e. the solute and solvent do not like to be together) then the
solubility is reduced, if it is higher (there is some positive interaction
between them such as donor/acceptor) then the solubility is increased. The
formula including all three terms is then:
Equ. 1‑11 Ln(x) = ΔF/R*(1/Tm – 1/T) + ΔCp/R [Tm/T – ln(Tm/T)-1]
– (ΔHmix -TΔSmix)/R [(1-x)²/T]
HSPiP allows you to play with these terms.
Clearly the dominant effect is still the melting point – the higher it is
(and the higher the enthalpy of fusion) the lower the solubility, but the
surprisingly large Cp effect and some assistance from a negative heat of mixing
can at least fight against the low solubility that a high MPt generally brings.
The fact that x is on both sides of the
equation for heat of mixing effects leads to some strange plots for high values
of ΔHmix. The
strange plots are not realistic because they happen to represent violations of
Gibbs phase rule. Whether they represent “oiling out” effects is a matter that
can be followed up by those who read the paper referred to above.
E-Book contents | HSP User's Forum
