Hansen Solubility Parameters in Practice (HSPiP) e-Book Contents
(How to buy HSPiP)
In earlier editions, this was a short
chapter. It was about adhesion and its main aim was to counter naive ideas that
adhesion was “all about surface energy”. The chapter has now expanded to bring
in some serious polymer physics of adhesion and has been extended to include
polymer solution viscosity and a return to basic polymer solubility. The logic
of combining them in one chapter will hopefully become clear.
On first reading you may well want to
abandon this chapter once the polymer physics starts to get difficult. But if
you come back to the chapter from time to time you’ll find that the concepts in
the later sections really throw a lot of light onto many of the issues you face
when using polymers and solvents. As is so common in science, insights from the
discussion of one problem (e.g. polymer adhesion) can be helpful in an
apparently unrelated area (e.g. the disturbing ability of polymer solutions to
become very viscous with small changes in conditions).
First let’s turn to the adhesion between
two polymer formulations. Whether you are interested in classical
polymer-to-polymer adhesion or getting an ink to stick to a polymer surface,
the considerations are the same.
Sticking
together
It turns out that HSP can provide insights
that help us distinguish between three types of adhesion. The first type of
adhesion, based on surface energy, is insignificant. The second, delightfully
named “nail adhesion” is quite common and (as those who prefer to screw things
together already know) quite unreliable. The third is the real adhesion for
those situations where things must stay stuck together – the wing on your
aeroplane or the legs on your chair.
Real
adhesion is not based on surface energies
There is a common misconception that
adhesion is all about surface energies. It is well known that a polymer of low
surface energy such as polyethylene has low adhesion, but once corona treated
has (a) a high surface energy which allows an applied coating to flow out
rather than bead up, and (b) good adhesion. From these facts the conclusion is
that the increased adhesion to the polyethylene is due to the high surface
energy.
There is a very simple disproof of this
view of adhesion. The surface energies of PVC film and PET film are identical.
Yet it is very easy to get printing inks to stick to PVC and very hard to get
them to stick to PET.
There is another common half-truth about
adhesion. It is well-known that roughened surfaces tend to have stronger
adhesion than smooth surfaces. The common explanation for this is that there is
more surface area for adhesion on the roughened surface. This is true, but the
effect is small. A typical “rough” surface like this probably has only 10%
extra surface area:
Figure 0‑1 A normal rough surface has a small increase in surface area

To double the surface area you need a surface of a dramatic
roughness (a high 1:1 aspect ratio):
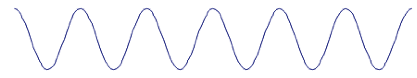
Figure 0‑2 It takes extreme roughness like this to double the surface area and
double the adhesion due to surface energy
The main reason why roughened surfaces
increase adhesion has to do with crack propagation. If there is a smooth
interface and a crack appears, all its energy can be transmitted straight along
the interface and the crack moves along swiftly, destroying adhesion. If there
is a rough interface, much of the crack energy is sent into directions away
from the crack so the crack does not propagate.
The myth of surface energy being important
for adhesion comes from some good, simple science. The work of adhesion Wa of
two surfaces can be simply calculated:
Equ. 0‑1 Wa= γ * (1 + Cosq)
Where γ is the surface energy and q is the contact angle of the second material with the first. If the
surface goes from wetting (q =
0º) to non-wetting (q =
90º) the work of adhesion changes only by a factor of 2. This is a small
effect when we’re talking about real-world adhesion where differences are
factors of 100’s.
The real explanation for the dramatic
increase of adhesion to polyethylene after corona treatment is the fact that
the polyethylene crystals are broken up at the surface and an open polymer
surface is available to interact with whatever you are trying to stick to the
surface. The meaning of “interact” is explained below. The functionalisation of
the surface by the treatment is a surprisingly small effect – often only
1 in 100 surface carbon atoms has been oxidised. This is enough to allow good
wetting of an applied coating (an important effect) but largely insignificant
in providing real adhesion
Before we come on to the key science of
real adhesion, it’s worth mentioning the Gecko effect. There have been many bad
explanations of how a gecko can climb up a smooth surface. It turns out to be
very simple. Van der Waals forces of two surfaces in intimate contact are more
than strong enough to support the weight of a gecko. So all the gecko needs to
do is make sure there is plenty of surface contact. The feet are designed with
multiple levels of compliance, ensuring that there is every chance for bits of
the feet to be in contact with as much of the surface as possible. In simulations,
it hardly makes much difference if the feet are high or low surface energy.
Going back to the issue of crack propagation and surface roughness, it’s clear
that cracks fail to propagate along the many interruptions in the surface
contact of the gecko.
But the key thing about gecko adhesion is
that it is perfectly useless for “real” adhesion. The gecko can lose adhesion
with a mere flick of its foot. And of course this is vital because the gecko
has to be able to walk. If adhesion were too good it would be stuck forever. So
gecko adhesion is closer to “scientific adhesion”. It’s elegant, functional,
explainable with simple surface physics, but ultimately useless for most areas
of adhesion that are of practical interest to humans. It’s common, for example,
to have layers of ink with good “gecko-like” adhesion but which peel off all
too easily when put under real-world test conditions.
We shall soon put some numbers to all this
which will show that surface-energy adhesions of ~0.1J/m2 are a factor of 100-1000
too small to explain real adhesion.
In what follows we largely ignore adhesion
of polymers to non-polymers such as metals. However, a later discussion about
chemical bonding between surfaces (of particular relevance to metal/polymer
bonding) shows that entanglement issues play an important and little-known
role.
HSP
to the rescue
When dealing with polymers, real-world
adhesion comes mostly from intermingling of polymer chains. The more the chains
are intermingled, the harder it is to pull them apart when stress is applied at
real-world speeds. It’s like untangling a ball of string. If you pull the
string very slowly and carefully it’s possible for it to slide out from the
tangles. But if you pull it quickly the tangles tighten upon themselves making it
impossible to move. In scientific terms if the polymers are above a “critical
entanglement limit” then adhesion is strong.
To get polymer chains to entangle (and stay
entangled – i.e. we’re ignoring temporary tricks such as solvents
compatibilizers) they have to be thermodynamically more stable when extended as
opposed to closed in upon themselves. If you try to mix two polymers with very
similar HSP (think of the extreme case where the HSP are identical because the
polymers are identical) then they will entangle readily (provided there is some
kinetic boost such as heat or solvent). Two dissimilar polymers will simply not
mix.
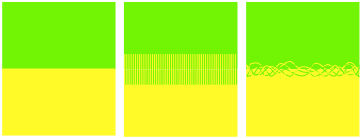
Figure 0‑3 No intermingling (surface energy only, poor adhesion ~0.1J/m2),
straight intermingling (“nail” adhesion ~1J/m2) contrasted with
entanglement (real adhesion 100-1000J/m2)
It’s obvious from the picture that the
polymer system on the left will have poor adhesion. The one in the middle has
good interpenetration of the polymers so you might think that the adhesion will
be OK – but it won’t be much good. We’ll see that it takes the situation
on the right with strong entanglement before decent adhesion is obtained.
The measure of HSP similarity between two
polymers is the HSP distance between them (taking into account their respective
radii). A short HSP distance is a strong indicator of the potential quality of
adhesion, providing they have a kinetic chance to mix via solvent or thermal
treatment. The effect of chain length is more difficult to predict. Short
chains will not have strong entanglement, but they are easy to intermingle.
Long chains will have very strong entanglement, provided they’ve had sufficient
exposure to solvent or heat. Similarly, linear chains are easier to entangle
but easier to disentangle than branched chains. And in a complex polymer, it
may happen that the average HSP is
not suitable for compatibility but certain segments (side-chains, blocks) are
more compatible, provided they are given the chance to come to the surface.
Sometimes it requires a compatible solvent to get the correct segments to flip
to the surface in order to bond with the other surface. But even though there
are many subtleties, the HSP distance is a crucial component of adhesive
thinking.
(We note in passing that these entanglement
issues are highly relevant to Environmental Stress Cracking, where higher
molecular weight variants/mixes of a polymer are often more resistant to ESC).
The HSP radii of the two polymers
encapsulate one other element of the problem. Polymers with small HSP radii
are, for various reasons such as high molecular weight or high crystallinity,
harder to access, so require a closer HSP match.
The polyethylene example now makes more
sense. Wetting and film retention alone does not assure acceptable adhesion, but
by breaking up the crystal structure through the corona treatment, interaction
with whatever you are trying to stick becomes possible. By adding some modest
(oxygenated) functionality, the HSP compatibility with many adherents is
increased. There’s a final sting in the tail about corona treatment for
increasing adhesion. The destruction of the surface which is a desirable
feature can be taken too far. You then have polymer rubble on the surface.
Adhesion of your ink to this rubble might be quite good. But it is building a
house on sand. It’s well-known, for example, that PET corona treatment can
create rubble and considerably reduce adhesion in many systems, making it a
difficult judgement call if you find that for your system the adhesion is
enhanced (once more, by breaking up the crystalline domain of bi-axially
oriented PET). How can you tell if that enhancement is permanent or something
that’s going to fail because the pile of rubble falls apart?
The
polymer physics of adhesion
The description above is OK as far as it
goes, but it’s a bit on the vague side. Let’s get deeper into the polymer
physics. For those who want a masterful modern summary of the physics, try the
review by Richard P.Wool, Adhesion at
polymer–polymer interfaces: a rigidity percolation approach, Comptes
Rendus Chimie 9 (2006) 25–44.
A useful starting point suggested by
Professor Wool is “nails” adhesion. His paper is a classic and well worth
reading: Richard P. Wool, David M. Bailey, Anthony D. Friend, The nail solution: adhesion at interfaces, J. Adhesion Sci. Technol., 10, 1996,
305-325. Take two blocks of wood and nail them together. The more nails N you
have, the longer, L, they are and the higher the friction µ between
the nail and the wood, the greater the adhesion. In fact the Fracture Energy
=0.5µNL2. If you do the calculation for typical short polymer
chains across an adhesive bond (as shown in the middle image of the diagram
above), the fracture energy turns out to be something like 1J/m2.
This is higher than any reasonable surface energy bonding energy of ~0.1J/m2.
But “real” adhesion is in the 100’s to 1000’s of J/m2, so “nails”
adhesion is rather close to useless. If you want two blocks of wood to really
stay together you use screws instead of nails. For polymers you can’t have
screws, but you can have entanglement.
There’s a precise description of
“entanglement” and it is therefore possible to calculate some “critical
entanglement parameters”.
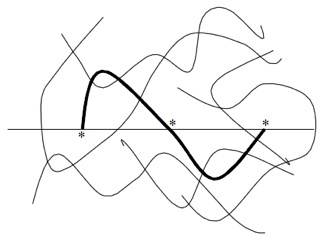
Figure 0‑4 The classic “Wool” diagram (with permission) showing the three
crossings necessary for entanglement.
A polymer chain can be said to be entangled
across an interface if it crosses it three times. One or two crossings are
equivalent to one or two nails, only three (or more) crossings ensure
entanglement.
It turns out there is a critical length
required for a polymer chain to be able to wander back and forth that many
times. This can be expressed as a critical molecular weight Mc or a
critical length Lc or critical “radius of gyration” Rgc (they are, of course,
inter-related). The formula for Mc and Lc are:
Equ. 0‑2 Mc=31(bz/C)2jM0C∞
Equ. 0‑3 Lc=bs(Mc/6M0)½
Unfortunately you need to know quite a lot
about your polymer before you can do the calculation. b is the average bond
length, bs (often confusingly shown as b) is the statistical or
“Kuhn” length, z is the number of monomers per unit cell of size C, j is the
number of bonds along a monomer (e.g. 2 for PS, PMMA, PE… and 12 for PC), M0
is the monomer molecular weight. C∞ is the “characteristic ratio”
for your polymer, a number that encapsulates the stiffness of the chain and
which varies between about 2-18.
A useful collection of these parameters can
be found in L. J. Fetters, D. J. Lohsey and R. H. Colby, Chain Dimensions and Entanglement Spacings, Physical Properties of
Polymers Handbook 2nd Edition, Chap. 25, 445-452, 2006.
Here are some typical values deduced from
that and other references:
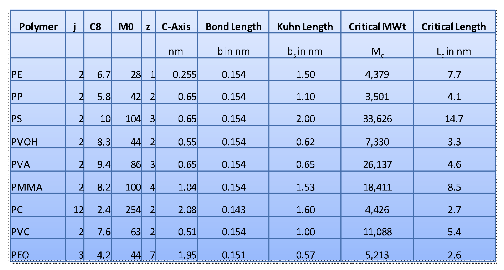
Figure 0‑5 Typical examples of the key parameters for entanglement
calculations.
When it comes to adhesion across a polymer
boundary these critical values are important. The two polymers form an
interlayer width, d. If d<Lc, the critical length, then you only
have nails adhesion. So the question arises, what is the typical value of d?
There is a well-known formula for this:
Equ. 0‑4 d=2b/(6χ)½
where b is the statistical bond length and
χ is the interaction parameter. It’s interesting to note that χ is sometimes
assumed to be a specific Flory-Huggins term, but in fact Hildebrand derived it
from pure solubility parameter considerations and it was later shown
(especially by Patterson) that (a) the Hildebrand and the Flory-Huggins χ’s are
essentially the same and (b) they both contain approximations that happily tend
to cancel out and give good results. Hansen took the Hildebrand definition and
put it into its now-familiar form:
Equ. 0‑5 χ=MVol/RT*((δD1-δD2)2
+ 0.25*(δP1-δP2)2 + 0.25*(δH1-δH2)2)
Now we have our χ it seems as though we can
easily predict adhesion from our HSP! Unfortunately it’s not that simple. There
is significant disagreement in the literature about the use of χ in these
calculations, not least because a standard way of measuring χ using Small Angle
Neutron Scattering (SANS) is itself open to much dispute.
Note that the MVol in the formula is the
molar volume of the polymer (not of the monomer unit). If you quadruple the
molecular weight then the MVol is quadrupled and the interlayer width is halved
(a 1/MVol½ relationship). This means that the laws of physics
are against you. As you increase the molecular weight to approach or exceed Lc,
the interlayer width decreases making it harder for d>Lc. That’s
one of the reasons it’s so hard to get really good adhesion between polymers.
Getting d≫Lc is important and the Number of Entanglements, Ne=d/Lc
is a very significant number. There are good experimental data showing that
bond strength is small when Ne<1 (i.e. below the critical
entanglement limit) and then has a linear dependence on Ne>1. So
if d=2Lc the bond strength is double that of d=Lc.
The Adhesion calculator in HSPiP can only
be seen as indicative. The χ calculation itself is uncertain, and you
have to enter estimates for many parameters to calculate d. Furthermore you are
calculating the interface for two polymers with only one bs and
MVol. And you are likely to be dealing with polymers of broad molecular weight
distributions rather than the idealised single molecular weight implicit in the
theory and used in the experiments that have proven the theory. But as a guide
to the sorts of issue you face when designing an adhesive interface it seems to
be much better than having no tool at all. What is certain is that a smaller
HSP distance will give better adhesion if the following three conditions are
met:
·
The molecular weights of both
polymers exceed Mc for critical entanglement
·
The calculated d>Lc
·
There has been sufficient
bonding time and mobility for the entanglement to take place
The last factor is the great unknown. If
you are melt-bonding two polymers then the time for them to diffuse to distance
d depends on t¾ MWt-7/2. The problem is that it’s
relatively easy for the polymers to diffuse by “Rouseian” dynamics but these,
by definition, do not involve entanglement, they just give you nail adhesion.
As you become entangled you are in the domain of Reptation dynamics (“a snake
sliding down a tube”) which are slower with a large dependence on MWt. The
“obvious” way to increase entanglement is to lower the viscosity via a good
solvent. Unfortunately, the more solvent that’s present, the further apart the
polymer chains are so the less likely they are to entangle. This is not to say
that solvent-based entanglement is bad, it’s simply to point out that polymer
physics are against you if you’re not careful.
A logical way to attain a good interface
between two polymers is found frequently in the world of real adhesives. A
di-block co-polymer made from the two polymers will obviously be compatible on
each side of the interface. The trick is to make sure that each block is long
enough to be firmly entangled within its own polymer, otherwise you have
di-block nail adhesion which is not as effective. The classic study on this
(Creton et al, Macromolecules, 25, 3075, 1992), using PS-PVP di-blocks showed a
fracture energy of ~5J/m2 when the PS was above its entanglement
length and the PVP was below it, with all the fracture failure on the PVP side.
Once the PVP exceeded its entanglement length the fracture energy rose to
~100J/m2.
Polymer
physics gets you again
In a paper Polymer-Solid Interface Connectivity and Adhesion: Design of a
Bio-based Pressure Sensitive Adhesive, most conveniently found online at http://www.che.udel.edu/pdf/WoolPaper.pdf
Wool and Bunker provide a startling demonstration of the importance of
entanglement. Suppose you have a surface (e.g. a metal) with functional groups
–Y and you want to bond a polymer with functional group –X which
forms a strong –X-Y- bond.
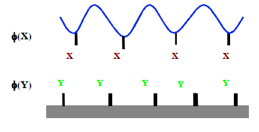
Figure 0‑6 What are the optimum φ(X), φ(Y) for good adhesion? Less than you
would think!
It seems obvious that the stronger the bond
and the larger the fraction of –X and –Y bonds (φ(X), φ(Y)), the
stronger the bond will be. “More” is obviously “better”. Yet Wool (and others)
have shown that a φ(X)>~1% leads to a rapid decrease in bond strength. How
can this be?
The key to this puzzle is the mode of
failure. Below this critical φ(X) the failure mode, not surprisingly, is
adhesive: the polymer pulls away from the surface. Above the critical value the
failure mode is cohesive: a thin layer of polymer remains on the surface. The reason
for this is entanglement, or, rather, loss of it. As you get more and stronger
–X-Y- links, the more the surface layer of polymer gets flattened onto
the surface and therefore the less it can
entangle with the rest of the polymer partly because of reduced mobility and
partly because the chains do not extend far enough into the bulk in order to
entangle. This is not a small effect. For their particular adhesive the
bond strength goes from 250 J/m2 at 1% to 50J/m2 at 1.2%,
a catastrophic reduction in practical adhesion for just a small increase in
φ(X).
There is no HSP message in this small
section – we simply find this to be a fascinating bit of science which is
surprisingly little-known.
A
quick way to test for contamination
All the adhesion science is worth nothing
if your surfaces are contaminated. So a quick way of testing for contamination
can be very handy. Although this chapter stresses that the common myth about
surface energy being important for adhesion is out by a factor of >1000,
surface energy tests can be an excellent way to identify contaminants. Although
one can use surface-energy pens (“Dynes pens”) there is an even easier way. It
was developed by the same Hansen who developed HSP and is an official Nordtest
Poly 176: (www.nordicinnovation.net/nordtestfiler/poly176.pdf
). A set of vials contain ethanol/water mixes varying from pure ethanol
(surface tension=22 dynes/cm) up to pure water (72 dynes/cm). Starting from the
ethanol end of the range, drops from each are placed on the test surface till
one is found that does not spread. That, then, is a guide to the surface energy
of that surface. This is not a way to
measure the surface energy, it is simply a guide. The point is that if you know
that you get a good bond with a surface that spreads drops up to 46 dynes/cm (10.9:89.1
ethanol:water) and your test piece spreads only up to 36 dynes/cm (23.4:76.6)
then there’s something wrong with your surface.
Hansen has used the technique to identify
contaminants on many items including wind-turbine blades and the cement lining
of a major tunnel. In both cases, just a few drops of ethanol/water were
sufficient to identify problems that could have had major long-term consequences.
Ethanol/water is not an aggressive solvent mix for most surfaces so the results
are seldom confused by surface damage from the solvent.
The test isn’t perfect. If your 46 dynes/cm
is covered by a 46 dynes/cm contaminant then you’ll never know. But very often
the contaminant will be some sort of lower surface energy oil and the test will
reliably detect it.
Polymer
viscosity
This section doesn’t have much to do with
adhesion. But because so many of the considerations are similar, and because
the calculations in the software are on the same modeller, we’ve included this
summary here. You may find the following a little frustrating. There seem to be
so many parameters and assumptions to hold on to and there seem to be many
uncertainties in the estimates. That seems to be the nature of this literature.
It is very difficult to find papers
that pin down all the key variables in one go and then link them to
experimental values. We’ve done our best. As ever, if the HSPiP community can
provide better ways of doing this, we’ll be happy to update this text and the
program.
First we need to define Rg, the
Radius of Gyration of the polymer. It is given by a formula that is similar to
the Mc calculation above. The C∞, j and M0 values are given for a range of polymers in the
software and b can be assumed to be 0.15nm if you don’t have a better value to
hand.
Equ. 0‑6 Rg0=b
[C∞Mj/(6M0)]½
Unfortunately, Rg depends on how
“happy” the polymer is within the solvent. This formula gives the Rg
for a polymer in a neutral solvent – i.e. one with neither positive nor
negative polymer/solvent interactions. Such a solvent is termed a theta solvent
and so we call this value Rg0.
As the solvent gets better, the polymer tends to expand so Rg
is larger. The traditional way is to say that Rg=Rg0 α2
where α is an expansion factor. Unfortunately there is little agreement on how
to know α. In HSP terms, polymer “happiness” is defined by the RED number. When
RED=1 the polymer is on the border between happy and unhappy – i.e. at or
near the theta point and the Rgo from the above formula is used. As
the RED reduces, the Rg increases (the polymer likes to be in the
solvent). Note that RED is in solubility parameter units. Chi and the
interaction coefficient are in energy units. A RED of 0.9 converts to a chi
effect of 0.81 by the squaring process. In other words the reduction in RED is
more important than a linear reduction. We’ve chosen a simple estimate of this
effect that works adequately with data from the classic known polymers. It’s
saying that for a good solvent, Rg goes as Mw1.6, for a
theta solvent it goes as Mw1.5 – but with a higher constant of
proportionality. Mw is the weight averaged molecular weight. For these sorts of
calculations, Mn, the number averaged molecular weight is not so useful.
Equ. 0‑7 RgRED=Rg
Mw(1.6 - 0.1 * RED) / Mw1.5 (1 + 1.5 * RED) / 2.5)
Because the viscosity effects are highly
dependent on Rg, we decided to let you decide which value to input. So we
output Rg0 and RgRED to give you an indication of likely
Rg values; you then use your scientific judgement in providing an
input value.
An alternative way to estimate Rg0
is to use handbook tables of Rg0/Mw½. We’ve
provided a selection of these values in the program. Obviously you need to
multiply them by Mw½ to get the Rg0 which you can
then compare to the value calculated in the program.
Figure 0‑7 The complexities of polymer adhesion and viscosity made as simple
as we possibly can
We must at this point take a side-track
into the connection between the definition of a theta solvent and RED number.
At first we hit an intellectual problem. A theta solvent corresponds to a RED=1
(Chi=0.5) where, by definition, we have no solubility. Yet polymer physicists
happily make solution measurements in theta solvents so there has to be some
solubility. The fix for this problem arises because the definition of a theta
solvent applies to a polymer of infinite molecular weight. As the molecular
weight gets smaller the polymer starts to have some solubility. In other words,
RED=1 for an infinite polymer means RED<1 for a finite polymer. The theory
of all this is discussed in the next section, but in short, a RED of 0.9 for a
polymer of MVol=10,000 in a solvent of MVol=100 corresponds to a theta solvent.
For a polymer of 100,000 the RED is 0.97. Given the many assumptions behind
these corrections, for the purposes of the viscosity calculations, a RED of 1
is assumed to be equivalent to a theta solvent, with the understanding that the
finite polymer is inside the border of the sphere and can therefore be
dissolved in sufficient amounts to make theta-solvent measurements. Errors in
other estimates far exceed the errors in this RED=1 approximation.
The viscosity of a dilute polymer solution
(we will define “dilute” in a moment) depends on the intrinsic viscosity [η]0
and the concentration. At very dilute
solutions the specific viscosity is given by [η]0C (Zimm), where specific
viscosity is (ηp- ηs)/ ηs and C is the
concentration (in the software C is in g/l). In other words, the specific
viscosity is the viscosity of the solution containing the polymer ηp
corrected for the viscosity of the solvent ηs. By extrapolating specific
viscosity to zero concentration you obtain [η]0. We can calculate [η]0
in units of l/g via:
Equ. 0‑8 [η]0=5
AvRg3/Mw
An alternative way to estimate [η]0
is to use handbook tables of K0 where [η]0 =K0Mw½,
where the “0” indicates that these are values for a theta solvent. Handbooks
may also provide K and a values for calculating [η] in good solvents where [η]
= KMwa and a is higher than the 0.5 for theta solvents (typically
0.6-0.7). It’s impractical to include these in HSPiP so use the given K0
values to cross-check (multiplying by Mw½) the intrinsic viscosity calculation
using the Rg for a theta solvent, then change to a good solvent and
assume that the enhanced Rg translates to a correctly enhanced [η].
Polymer chains start to interact even in
dilute solutions. Because of this the general “dilute solution” formula for the
traditional way of reporting polymer viscosities, ηp/ ηs is
given by the following formula. For
simplicity the ratio ηp/ ηs will be called η in the
following;
Equ. 0‑9 ηdilute=1+[η]C+0.4([η]C)2
Above a certain critical concentration, C*,
the solution ceases to be “dilute” and Rousean dynamics take over and the
(hypothetical) Rousean viscosity is given by:
Equ. 0‑10 ηRouse=
([η]C)1.3
C* is the point at which the
polymer “blobs” start bumping into each other. It is calculated as
Equ. 0‑11 C*=Mw/(AvRg3)
Where Av is Avogadro’s number (6.022E23).
The Rousean domain doesn’t last long. As
the concentration increases, the polymers start to get entangled and viscosity
starts to rise dramatically. This critical concentration depends on Ne (the
number of monomer units in a critical entanglement molecular weight Me).
Ne= Me/M0, where Me is 1/3 the Mc
we’ve calculated above. So:
Equ. 0‑12 Ne=(Mc/3)/M0
Knowing Ne we can then calculate
the critical entanglement concentration, Ce. We called the Rousean
domain hypothetical because the entanglement effects start to kick in fairly
quickly (at C>0.5Ce) and it seems hard to spot true Rousean
viscosity because at 0.5Ce you can be close to the dilute limit.
Equ. 0‑13 Ce=Ne(3v-1)/(6.6/C*)
The 3v-1 term would be a simple 0.5 in a
theta solvent where the polymer/solvent interactions are neutral. But as you go
into good solvents it gets easier to entangle. For classic polymers in good
solvents, the exponent term, v ~0.585, making 3v-1=0.755, a big increase on
0.5. As a practical heuristic we’ve proposed the following for v:
Equ. 0‑14 v=0.585-(0.085*RED)
With all these equations we can now predict
viscosity over the entire range of polymer concentrations. If you play with the
Viscosity modeller you will find that small changes in v or in Rg can make
large changes to the calculated viscosity. This makes it very hard to be sure
that you’ve got good predictions. You will also notice that the three graphs
plotted (dilute, Rousean, entangled) don’t overlap. This is because no entirely
self-consistent description of all three domains seems to exist and in any case
the Rousean curve is hypothetical. The point of the graph is to show the general
form of typical polymer viscosity behaviour.
If you find this disconcerting, the reality
of polymer solutions is that relatively small changes (molecular weight and/or
its distribution, concentration, temperature, solvent blend) can produce
relatively large changes in viscosity, so the errors apparent in the lack of
overlaps are likely to be smaller than the effects arising from, say, molecular
weight distribution.
If you start going to high RED numbers
(>1) and large concentrations you will find that the calculations become
decidedly odd. But remember, at RED>1 you cannot get high concentrations, so
the calculations are irrelevant!
There’s one more key fact. The viscosity
we’ve calculated after all this hard work is only the zero-shear viscosity.
Predicting the non-Newtonian changes in viscosity at higher shear is, perhaps,
a challenge for future editions of HSPiP.
But the point of this section is not to provide a 100% reliable viscosity
predictor (as far as we know such a thing doesn’t exist). Instead we’re giving
you a modeller where you can see the general effects of molecular weight,
solvent compatibility (RED number), concentration etc. and therefore better
understand what’s going on in your own system. If you are already an expert in
the field, then this section won’t be necessary. If you are not an expert then
hopefully the calculations and graphs will provide you with some insights that
will help you better understand the literature of polymer viscosity and apply
it to your own polymer/solvent system.
Because we’ve taken a giant side-track into
polymer physics, we might as well finish off with some thermodynamics that tidy
up some loose ends in the explanation of the polymer sphere radius.
Really
understanding polymer solubility
In the “minimum theory” chapter we
described how solubility was about thermodynamics. But we didn’t give a
thermodynamic formula for this. It’s now time that we did. We use the
convention that “1” refers to the solvent and “2” the polymer and we use V1
and V2 for the molar volumes and ϕ1 and ϕ2
for the volume fractions. It’s more normal in thermodynamics to use mole
fractions, but because of the huge disparity in molar volumes, the numbers
don’t look very helpful. Because of this disparity the “x factor” is
introduced. This is simply x= V2/V1.
So now we can introduce the formula for the
partial Gibbs free energy of the
solvent on dissolving a polymer:
Equ. 0‑15 ΔḠ
1=RT [ln(ϕ1) + ϕ2(1-1/x) + Distance2
ϕ22 V1/(4RT)]
The first two terms are the entropy of
mixing, the third term is the enthalpy of mixing. The factor of 4 is there
because of the factor of 4 in the (δD term
of the) HSP Distance calculation. The combination of the first two terms is
usually called the Flory-Huggins entropy.
Because negative free energies are good for
solubility, it’s important to note that the 2nd and 3rd
terms are both positive. So solution is only possible because of the ln(ϕ1)
term. As you dissolve more polymer (ϕ2 increasing), ϕ1
decreases so ln(ϕ1) gets more negative – the classic
entropic driving force. But of course the ϕ2 term gets ever
larger, fighting against solution. Only if x≤1 does the ϕ2
term cease to be a problem. But x is always large so the 1-1/x term is always
>0. For an infinite molecular weight polymer, 1/x=0 so the ϕ2
effect is maximum. As the polymer gets smaller the ϕ2 effect
reduces (though not by a lot) and the chance of being soluble increases.
Non-intuitively, a larger solvent means a reduced ϕ2 effect, so
in this respect small solvents are bad.
The Distance2 factor in the
third term reminds us that the smaller the HSP distance, the higher the chance
of dissolving. The sphere radius then comes from when the Distance is large
enough for the 3rd term to produce a positive ΔḠ. As we will
shortly see, this distance is between 7 and 8, which is what is found
experimentally for a large number of polymers. The reasons for variations
around this number will shortly emerge. And because the 3rd term
also includes V1, we will soon see that small solvents are nearly
always a good thing, as intuition and practical experience shows. In other
words, the 3rd term solvent size effect (small is good) usually
outweighs the ϕ2 effect (small is bad).
It’s not obvious how all these terms work
out in practice. So we’ve made a Polymer Solution Theory modeller that lets you
input the key properties: V1, V2 and Distance. The ΔḠ
is then plotted over a ϕ2 range of 0-0.3. The scale for ΔḠ
is deliberately made small so that the details can be seen around the important
0 line. In particular, the classic case of a 10,000:100 polymer:solvent gives
an inflexion point at a critical value which translates to a Chi value of 0.605.
For an infinite polymer, this point of inflexion moves to 0.5, which is why a
Chi of 0.5 is seen as the definition of borderline solubility (theta solvent).
Because of the relationship between Chi and Distance for an infinite polymer a
Distance of 7.05 provides the boundary of solubility. This can be calculated
from first principles, but you can show this for yourself using the modeller.
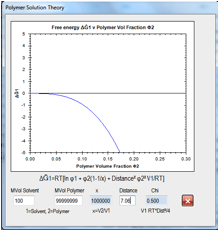
Figure 0‑8 For an “infinite” MVol polymer the thermodynamics of solubility go
critical at Chi=0.5 or a Distance of 7.06.
For the 10,000 polymer (as shown in the
figure below) it’s a distance of 7.77. The ratio 7.06/7.77 ~ 0.9, which is why
we stated above that a RED of 0.9 suffices for a real polymer compared to an
infinite polymer.
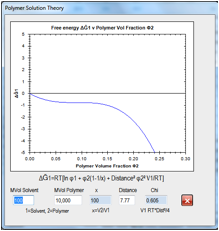
Figure 0‑9 For a 10,000 MVol polymer in a 100 MVol solvent, the thermodynamics
of solubility go critical at Chi=0.605 or a Distance of 7.77. That’s why we
often say that a typical polymer HSP radius is 7-8.
Remember, this is all theory based on many
assumptions. It is well-known, for example, that the Flory-Huggins entropy is
not a perfect approximation. Real solvents and polymers can’t be calculated
with this precision and some spheres have a radius of 10 and some of 5.
Let’s first see why we might have a small
radius. There’s an important term missing from the ΔḠ equation. If the
polymer is crystalline then (thermodynamically speaking) before it can dissolve
it first has to melt. This “crystalline” energy (sometimes confusingly called “ideal
solubility”) is discussed in the Predictions chapter but the formula is simple:
Equ. 0‑16 C = ΔF/R*(1/Tm – 1/T)
where ΔF is the enthalpy of
fusion and Tm is the melting point. For normal crystalline solids
these terms are readily known. For polymers things are seldom so crystal clear.
We are therefore making no attempt to predict it. However, what is obvious is
that the larger the value of C the smaller the Distance needed to ensure
solubility. That is why highly crystalline polymers have such small radii.
Polyethylene is a classic example. Its HSP mean that finding a compatible
solvent is not at all hard. But solution experiments using polyethylene
normally take place at ≫100ºC so that
the (1/Tm – 1/T) term becomes small enough to allow the
solvent to dissolve the polymer. The reason that you find solvents such as
Decalin being used for polyethylene is not because they have special solubility
parameters, but because they have a high enough boiling point to be able to do
experiments at 130ºC.
Rather satisfyingly we can come back to the
corona treatment of polyethylene. The disruption to the crystallinity at the
surface means that the C term becomes small so that ordinary solvents at low
temperature have a chance of interacting strongly with the top few nm of the
polymer film, provided their Distance from (perhaps slightly functionalised)
polyethylene is not too large.
One possible reason for a large radius
comes from the following inverted reasoning.
Backward
thinking
It’s obvious that if the polymer isn’t
soluble in the solvent then the solvent isn’t soluble in the polymer. This
“obvious” fact is completely wrong. The “x-factor” above explains why. If the 1
and 2 of solvent and polymer are swapped then the thermodynamics stay the same
but the important ϕ2 (1-1/x) term is transformed. For a 10,000
polymer and the same solvent, x is now 0.01 so the ϕ2 term is
multiplied by 1-100 which is decidedly negative, so this term strongly aids
solution.
In other words, even though the polymer may
be hardly soluble in the solvent, the solvent may be highly soluble in the
polymer. This very important point is often missed.
For example, it illuminates the discussions
on diffusion into polymers. Even though the polymer may never dissolve in the
solvent, there is still a chance that the solvent will dissolve in the surface
of the polymer, starting the diffusion process. The HSP distance is still very
important – the smaller the distance the larger the amount of solvent
that will dissolve in the polymer – even though classic HSP solution
experiments might show that the polymer is effectively insoluble in the
solvent. You can (as discussed in later chapters) determine HSP via
diffusion/permeation experiments. Solvents that are no good for dissolving the
polymer can still permeate reasonably, so the sphere based on these experiments
can be large.
This effect also explains why the HSP
spheres of solvent swelling experiments can be larger than spheres of true
polymer solution.
This doesn’t mean that the
diffusion/permeation or swelling spheres are “wrong”. You as the scientist
choose your definition of the sphere according to your purpose. If you are
interested in polymer diffusion/permeation or polymer swelling then such a large
sphere is correct. You only go scientifically wrong if you try to use the
numbers from one domain (swelling) in another (polymer dissolving in the
solvent).
This backward thinking isn’t just for
polymers. A classic example is that the solubility of water in hydrocarbons is
hardly affected by the MVol of the hydrocarbons. Yet the solubility of hydrocarbons in
water is highly MVol dependent. The best way to understand this is via the
classic solubility equation. At first there seems some confusion as the
Distance term now contains V2, the solute, whereas in the polymer
case it was V1. The reason is that in the polymer case we were
calculating the partial Gibbs free energy of solution, with the focus on V1.
In the classic solubility case we are calculating the activity of the solute,
so the focus is on V2.
Equ. 0‑17 ln(a2)=ln(mf2)
+ Distance2 ϕ12 V2/(4RT)]
Here we have the activity of solute 2 in
solvent 1. We use mf2 for mole fraction rather than the conventional
“x” to avoid confusion with the use of x above.
When we are trying to dissolve water in
hydrocarbons, V2 is that of the water and because Distance2
doesn’t vary all that much between hydrocarbons, solubility does not change
much.
When we try to dissolve hydrocarbons in
water, V2 is that of the hydrocarbons and the activity increases
(solubility decreases) as they get larger.
Finally, although in the case of water
dissolving in hydrocarbons MVol makes little difference, we need to return to
the general rule that smaller solvents are better for polymers.
This can easily be checked from the Polymer
Solubility Theory modeller. Just change MVol Solvent from 100 to 90 (whilst
keeping the HSP Distance the same) and a situation of critical solubility becomes
one of good solubility:
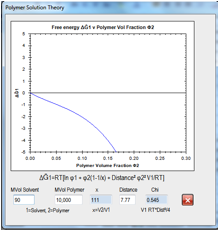
Figure 0‑10 For the same HSP Distance, if the solvent MVol decreases, the free
energy curve ceases to be critical and the polymer is likely to dissolve.
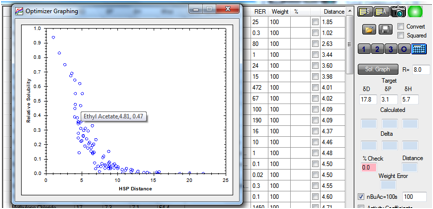
The Solvent Optimizer contains a simplified
version of the solubility/distance equation. If you enter the Radius of your
polymer, the “Relative Solubility” is calculated as exp(-Distance2 V2/Rterm)
where we’ve assumed a constant ϕ12 and use an
“Rterm” which is proportional to the chosen Radius (and contains the RT term).
A larger Radius means a smaller negative exponential and therefore a larger
Relative Solubility. With the mouse you can read off the various solvent values
from the graph.
Summary
Congratulations if you’ve reached this far.
Our view of this chapter is that it poses three further challenges.
1. The “easy” challenge lies in
understanding the formulae and getting used to playing with the modellers in
order to work out what’s going on. We’ve peppered the modellers with the
formulae so that when you get a surprise (as we often do) you can work out why
changing parameter A affects parameter B which in turn affects the output C in
a way you didn’t expect.
2. The “hard” challenge is to find ways to
map the theory into one’s scientific and technical work. Even though the theory
is complex enough, it still contains lots of simplifying assumptions and, as we
admit, only gives “indicative” numbers. Yet we’ve found in our own real-world
work that the modellers and the ideas behind them have proved crucial in
disentangling (pun intended) complex issues. They are particularly good in
providing a common numerical language that a cross-functional team with varying
expertise in physics, chemistry and analytical science can use to build a
fuller picture of what’s going on in their system.
3. The final challenge is for ourselves,
with the help of the HSP community, to build further on these foundations and
increase the range and depth of the predictive power derived from the theory.
E-Book contents | HSP User's Forum
