Molecular Orbital & Properties: TSL: MOPAC calculation
A problem when building this transition state database by the difference between MOPAC7 and MOPAC97.
MOPAC will optimize structure by a XYZ coordinate system, if XYZ is specified by a keyword. It seems to be easy to arrive at the transition state with righter searching MOPAC97 for a transition state which specified XYZ. When it calculates by specifying this keyword XYZ, although XYZ is contained into the keyword for the output of the summary file of a calculation result, since the structure itself is an internal coordinate system, cautions are required of MOPAC7. That is, if it tries to read test.arc even if XYZ is specified, it will break out that the structures differ by the XYZ coordinate system, an internal coordinate system, or MOPAC7 and MOPAC97. In this database, the keyword XYZ of the summary file after calculating by XYZ specification by MOPAC7 deleted.
- IONIZATION POTENTIAL = 9.665508 EV FINAL GEOMETRY OBTAINED CHARGE - |
MOLECULAR WEIGHT = 140.161 FINAL GEOMETRY OBTAINED CHARGE |
Only one imaginary frequency appears in a transition state. The first lump was recorded on the database in as a result of oscillating calculation (keyword FREQ) of MOPAC. How to attach the title of oscillating calculation by MOPAC7 and MOPAC97 differs.
MOPAC7 ROOT NO. 1 2 3 4 5 6 |
MOPAC97 Root No. 1 2 3 4 5 6 7 8 |
Moreover, MOPAC97 are eight vibration although lumps are six vibration in MOPAC7.
In MOPAC7, "1 (new-line)" appears in some places. although seemingly this is the vestiges when changing a page by a printer -- this -- an obstacle -- since it was stinking, it eliminated.
MOPAC7 36 0.16423 0.06577 -0.02412 -0.06345 0.03391 -0.00448 |
Other versions have not used.
Please inform me if you know other versions.
Trouble when calculate transition state
When looking for the transition state of vinyl acetate and vinylidene chloride, a trouble just for a moment ... When searching for a transition state by MOPAC, it meets well "A trouble and a solution"
Although MOPAC7 does not know, in MOPAC97, the number of times of geometry optimization is 500 times. When it is going to search for a transition state,
MOPAC 7 CYCLE: 739 TIME: 0.00 TIME LEFT: 7096.0 GRAD.: 9.576 HEAT:-63.95119 |
MOPAC 97 CYCLE: 497 TIME: 0.000 TIME LEFT: 55.95M GRAD.: 0.103 HEAT:-64.24002 |
It may become and calculation may stop at MOPAC97 (calculation is completed in MOPA7). When such, it is in a keyword.
CYCLES=2000
It puts in. They are vestiges when calculation of MOPAC has taken many days.
But as for this VAc-VDC, a transition state can be found. If oscillating calculation is performed using the structure of the transition state,
| A FAILURE HAS OCCURRED, TREAT RESULTS WITH CAUTION!! |
A SCF CALCULATION FAILED error will come out and stop. Although this may have truly bad structure and SCF may not be found,
ITRY=2000
When the number of times of SCF was increased, calculation was completed with no problems in this case, and it carried out perfectly. A transition state can be found.
In addition, when the calculation which searches for a transition state goes wrong.
0. Change the structure of a start.
(In this case, it makes, for example from VAc-AN.)
1. Replace an internal coordinate system and a XYZ coordinate system.
2. Replace and calculate MOPAC7 and MOPAC97.
3. Move and calculate a part of molecule.
When TS keyword of MOPAC departs from a transition state library like this time by a fairly powerful keyword, the transition state right quite comfortably can be found.
Although the transition state was able to be found, when oscillating calculation goes wrong.
NORMAL COORDINATE ANALYSIS Root No. 1 2 3 4 5 6 7 8 |
For example, such only one imaginary vibration, a solution may be found. Shortly after looking at vibration of this imaginary, it understands, but this is not going in the reaction direction. (Vibration of a twist) Since vibration of imaginary becomes smaller than -200 in case of the transition state right on experience of it, the larger thing than it suspected and has started.
In addition, when two or more vibration of imaginary exists, it is calculation failure clearly.
0. Change the structure of a start.
1. Move and calculate a part of molecule.
These techniques are effective. It seems that TS keyword of MOPAC97 is powerful compared with the thing of MOPAC7, and two or more vibration of imaginary does not exist probably.
If it is the usual calculation, when searching for the first
transition state
PM3 PRECISE TS UHF (XYZ)
then
PM3 PRECISE FORCE UHF (XYZ)
Only vibration is calculated by doing. When carrying out this FORCE calculation
very occasionally, structure may also move. Let's confirm whether filename.arc
(FOR012) etc. is made to the calculation result. In MOPAC7, since like,
once [ when such a tendency is especially strong ] calculating TS according
to the case, after replacing XYZ and internal coordinates and calculating
TS several times, FORCE calculation is performed.
By calculating by the keyword of PM3 PRECISE TS UHF (XYZ) first, in MOPAC7, if FOR012 is made, the portion on it is eliminated, and if TS is calculated by changing it into FORCE, oscillating calculation can be performed. That is, even if the charge is contained in input data, it is calculable satisfactory. It seems that however, the portion of this charge cannot be calculated in MOPAC97 unless it eliminates. It is troublesome to eliminate the portions of a charge one by one. If the Search button is pushed without choosing a radical and a monomer by extraction of structure , empty text area will appear. Only the portion of the structure of a molecule is copied there (every value of a charge), and the Read button is pushed. Probably, it will be good in calculating MOPAC by copying it to a text editor etc., and rewriting and saving TS to FORCE, since the structure where the charge was eliminated will be acquired if the GetStructure button is pushed immediately after that. The meaning which checks the structure of TS is also effective.
The cautions when changing an atomic kind. When changing an atomic kind, the remaining portion is moved using the standard joint length of the new atom. Under the present circumstances, in a transition state, although many are so, since there is no combination between a radical and a monomer, the phenomenon in which it does not move will come out of one of molecules. Even when it is troublesome, in case a molecule is Edit, please make combination between a radical and a monomer. (When changing the kind of some atoms of a ring, joint length does not change.)
Please put in a MMOK keyword, in calculating amide.
If many MOPAC97 (thing of attachment in Chem3D) of the Mac version is calculated continuously
Not Enough Time to Complete Hessian
The message to say will come out and stop.
Then, application is ended and it rerises.
(I think that it is a mere bug.) It seems that cooperation with Chem3D is also amusing, calculation of MOPAC97 is also slow and offset values, such as the number of times of SCF, are not suitable for it, either, although it is its personal opinion.
Merit that's right direction it seldom uses.
Since there is the restriction [ atomic number / of MOPAC7 which he is using / maximum ] 40, the thing exceeding it is using MOPAC97 reluctantly.
In the keyword at that time
T=100000 CYCLES=20000 ITRY = 2000 is added.
When the coordinates which JAVA generates are close to zero, the account of a table of reference may become like 1.23xxxxE-4. Since it will become an error if MOPAC is run as it is (error which says that the atom E does not exist), please rewrite with 0.000123xxx manually.
MOPAC PM3
MOPAC PM3 conclusion
Activation energy = Heat of Formation of a transition state - (HF of a monomer + radical HF)
|
AAR |
AllCR |
AMR |
AMDR |
ANR |
BDR |
cHnR |
CHOR |
GMAR |
MAAR |
| AA |
7.32 |
6.60 |
7.23 |
6.70 |
7.41 |
11.08 |
9.32 |
7.19 |
10.63 |
11.18 |
| AllC |
8.65 |
7.67 |
8.61 |
7.86 |
8.44 |
12.05 |
11.03 |
8.54 |
11.36 |
11.25 |
| AM |
9.60 |
8.91 |
9.52 |
9.18 |
9.65 |
13.40 |
11.72 |
9.57 |
13.39 |
13.74 |
| AMD |
9.07 |
8.74 |
9.38 |
8.79 |
9.25 |
13.66 |
11.68 |
9.10 |
13.50 |
13.71 |
| AN |
9.07 |
8.84 |
9.03 |
8.47 |
9.28 |
12.72 |
10.58 |
8.66 |
12.54 |
12.62 |
| BD |
5.95 |
6.26 |
5.96 |
6.14 |
6.23 |
10.01 |
8.25 |
5.83 |
9.35 |
9.24 |
| cHn |
11.30 |
11.11 |
11.39 |
11.82 |
12.41 |
18.01 |
11.91 |
11.52 |
15.92 |
15.78 |
| CHO |
9.18 |
8.69 |
9.12 |
8.94 |
9.35 |
13.23 |
11.62 |
9.00 |
13.14 |
13.34 |
| GMA |
6.95 |
7.20 |
6.89 |
6.72 |
6.25 |
11.81 |
8.90 |
6.90 |
9.91 |
10.35 |
| MAA |
8.50 |
7.90 |
8.44 |
8.28 |
7.87 |
12.29 |
10.04 |
8.26 |
13.46 |
12.02 |
| Mal |
11.23 |
10.45 |
12.56 |
10.25 |
12.07 |
13.45 |
11.44 |
10.94 |
15.87 |
14.97 |
| MAN |
7.57 |
7.48 |
7.62 |
7.14 |
7.09 |
11.31 |
9.32 |
7.24 |
10.70 |
10.86 |
| MeO |
7.38 |
8.22 |
8.45 |
7.91 |
7.84 |
12.77 |
10.79 |
7.63 |
11.19 |
11.20 |
| MMA |
8.75 |
7.57 |
8.85 |
8.91 |
7.75 |
12.72 |
11.12 |
8.84 |
12.12 |
12.15 |
| St |
5.97 |
6.10 |
5.98 |
6.05 |
6.02 |
9.90 |
5.38 |
5.91 |
9.25 |
9.22 |
| Vac |
6.82 |
7.27 |
6.82 |
6.31 |
6.82 |
11.68 |
10.09 |
6.83 |
10.41 |
10.36 |
| VC |
8.32 |
8.57 |
8.39 |
8.46 |
8.75 |
12.78 |
10.72 |
8.41 |
12.15 |
11.93 |
| VDC |
7.59 |
7.47 |
7.45 |
7.56 |
7.72 |
|
|
7.37 |
11.02 |
11.05 |
| VDF |
8.22 |
|
8.13 |
8.04 |
8.49 |
|
|
8.08 |
11.38 |
11.52 |
| VF |
8.48 |
8.65 |
8.44 |
8.47 |
8.71 |
12.78 |
10.86 |
8.51 |
11.86 |
11.91 |
|
|
|
|
|
|
|
|
|
|
|
|
MalR |
MANR |
MeOR |
MMAR |
StR |
VacR |
VCR |
VDCR |
VDFR |
VFR |
| AA |
6.23 |
11.07 |
7.23 |
11.62 |
9.88 |
6.59 |
6.60 |
10.41 |
4.87 |
5.45 |
| AllC |
7.12 |
11.82 |
7.84 |
11.63 |
12.24 |
8.27 |
7.51 |
10.93 |
4.54 |
6.29 |
| AM |
9.37 |
14.69 |
9.84 |
13.92 |
12.41 |
9.15 |
9.00 |
12.70 |
7.33 |
7.95 |
| AMD |
7.42 |
13.80 |
9.94 |
14.09 |
12.54 |
9.03 |
8.70 |
12.31 |
5.87 |
7.26 |
| AN |
8.29 |
12.70 |
7.39 |
13.03 |
11.62 |
7.56 |
8.47 |
11.71 |
5.22 |
6.51 |
| BD |
4.94 |
9.25 |
6.57 |
9.66 |
9.40 |
7.20 |
6.03 |
8.93 |
|
4.21 |
| cHn |
9.09 |
15.69 |
11.51 |
16.27 |
17.07 |
11.67 |
12.91 |
14.34 |
7.51 |
10.00 |
| CHO |
7.76 |
14.38 |
8.11 |
13.69 |
12.23 |
7.61 |
8.78 |
12.23 |
5.41 |
6.50 |
| GMA |
5.97 |
11.57 |
7.75 |
10.73 |
9.57 |
6.17 |
8.14 |
10.82 |
3.16 |
4.39 |
| MAA |
7.00 |
13.24 |
8.35 |
12.39 |
10.50 |
7.77 |
8.08 |
11.89 |
5.88 |
6.45 |
| Mal |
10.84 |
15.93 |
7.47 |
15.13 |
12.55 |
7.31 |
9.84 |
13.66 |
7.96 |
7.91 |
| MAN |
6.72 |
10.72 |
6.29 |
11.23 |
9.44 |
6.61 |
7.16 |
10.54 |
4.12 |
5.31 |
| MeO |
5.27 |
11.29 |
7.35 |
11.68 |
11.79 |
7.71 |
8.08 |
10.48 |
4.15 |
5.77 |
| MMA |
6.93 |
11.84 |
8.62 |
12.70 |
10.93 |
8.07 |
8.36 |
12.06 |
6.00 |
6.61 |
| St |
4.56 |
9.41 |
5.57 |
9.79 |
9.19 |
5.71 |
6.05 |
9.21 |
2.63 |
4.07 |
| Vac |
5.21 |
10.50 |
6.34 |
10.94 |
10.52 |
6.53 |
6.98 |
11.20 |
3.28 |
5.07 |
| VC |
7.29 |
12.00 |
8.05 |
12.32 |
12.14 |
8.03 |
8.21 |
11.27 |
4.62 |
6.29 |
| VDC |
7.01 |
|
6.42 |
11.41 |
10.50 |
5.55 |
7.23 |
10.54 |
4.38 |
5.44 |
| VDF |
7.95 |
|
7.25 |
11.84 |
11.05 |
|
7.74 |
11.28 |
4.46 |
5.86 |
| VF |
7.12 |
11.99 |
7.80 |
12.29 |
11.94 |
7.79 |
8.26 |
11.16 |
4.48 |
6.30 |
For the moment, nine transition states cannot be found. These all returned the error of not having reached SCF and have stopped. In this case, the result was the same even if it enlarged the value of ITRY. This error of characteristic one takes place easily at the time of the monomer (radical) of the symmetry of vinylidene chloride and vinylidene fluoride. When , such as Allyl chloride, butadiene, and cyclohexene, ( when reactivity is scarce) radical, I feel that this phenomenon has occurred for the radical at that time.
For example, if a monomer called cyclohexene regards as horizontally and goes, it needs a high value for activation energy with everybody 11kcal /mol or more. A Mal radical, a VDF radical, etc. may be able to react barely. It seems that it reacts and the made cyclohexene radical (cHnR) can react well with a styrene monomer, butadiene, GMA, etc. Although a styrene monomer reacts very easily, a styrene radical cannot react easily. This is explained to be because for the styrene radical to be stabilized by resonance. It says since what vinyl acetate (VAc) generally cannot receive a radical attack in easily has the unstable generated radical. But the calculation result of MOPAC has brought a result that VAc monomer react with some type of radicals . On the other hand, the reactivity of a Vinyl Acetate radical (VAcR) is high. The radical which will be generated once it opens, although it is hard to open double bond of VAc is very unstable, it is rich in reactivity, and it is explained that it has very high homopolymerization velocity.
I think that this talk will shift greatly when making an electron donor acceptor complex from the talk applicable to Free Radical Polymerization and reacting.
The reaction of such a radical and a monomer A textbook explains as follows.
Radical polymerization theory
"radical-polymerization theory" (Imoto P61- Japanese Polymer Text Book)
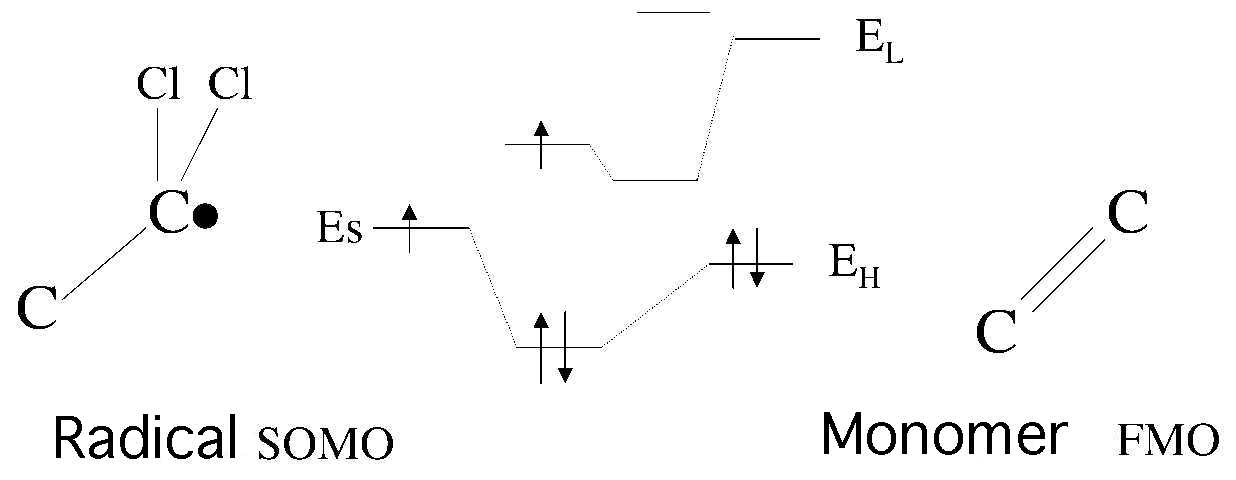
Radical SOMO, HOMO of a monomer, and orbital mixture start.
Next, the anti-bonding orbital then made causes LUMO of a monomer, and orbital mixture.
Then, I think that a natural thing thinks that dE can be estimated from radical
SOMO, HOMO and LUMO of a monomer.
Then, make such a table.
name |
dE |
RaSOMO |
MonHOMO |
MonLUMO |
MAA-Mal |
14.97 |
-9.682 |
-11.708 |
-1.548 |
AN-VDC |
7.72 |
-9.855 |
-9.744 |
0.327 |
AA-AMD |
12.31 |
-8.803 |
-9.794 |
0.143 |
AN-AM |
9.65 |
-9.855 |
-11.067 |
-0.078 |
MAA-AA |
11.18 |
-9.682 |
-11.147 |
-0.17 |
AN-AN |
9.28 |
-9.855 |
-10.886 |
-0.187 |
CHO-VDF |
8.08 |
-9.93 |
-10.759 |
0.227 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vac-cHn |
11.67 |
-8.785 |
-9.593 |
1.168 |
MeO-MeO |
7.35 |
-8.352 |
-9.515 |
1.302 |
MMA-MeO |
11.68 |
-9.566 |
-9.515 |
1.302 |
VC-AllC |
7.51 |
-8.928 |
-10.269 |
0.621 |
MAA-Vac |
10.36 |
-9.682 |
-10.083 |
0.687 |
BD-AA |
11.08 |
-8.848 |
-11.147 |
-0.17 |
First of all, calculate this by multiple regression. Since a correlation coefficient is 0.29, there is almost no correlation.
Formula
dE=0.963*RaSOMO-1.099*MonHOMO+0.621*MonLUMO+6.852
It comes to be alike. This is the plot.
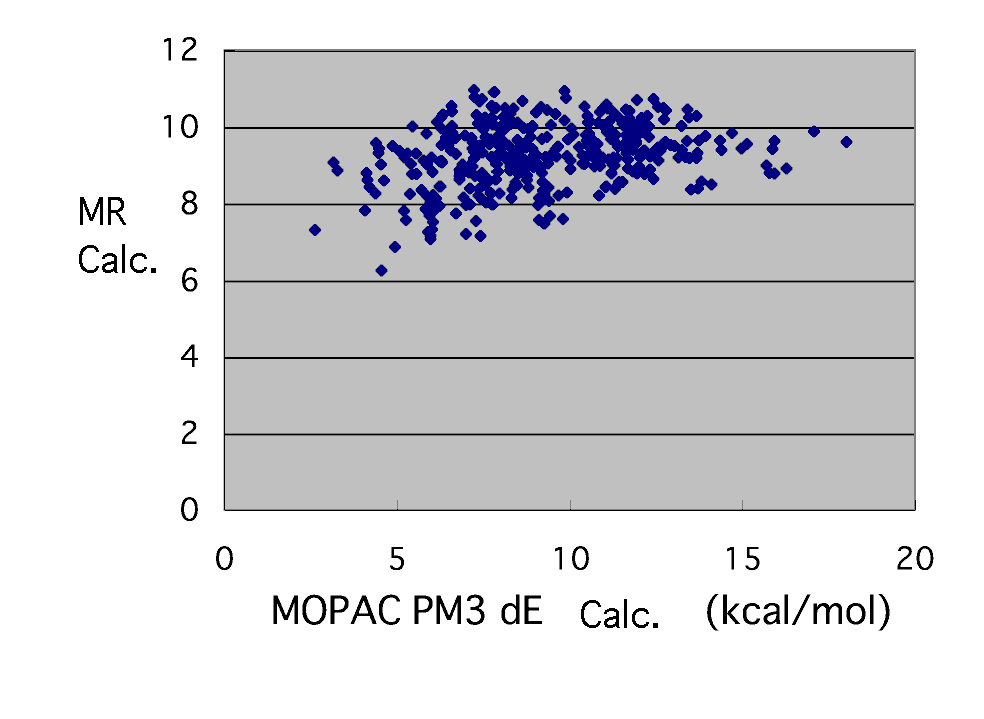
In a correlation coefficient 0.29, it is useless truly.
If considering why being useless radical activity is high (RaSOMO being
large), stable and low reactant monomer (MonHOMO -- small) can react well.
Probably,
it will be natural that accuracy does not come out, since all
of such an interaction are not taken into consideration in multiple regression.
Then, analyze by the neural network in consideration of the interaction among
the inputs.
The following results were brought, when the number of the middle layer neuron was set to 3 and it learned 715500 times with the bias neuron (reconstruction learning method).
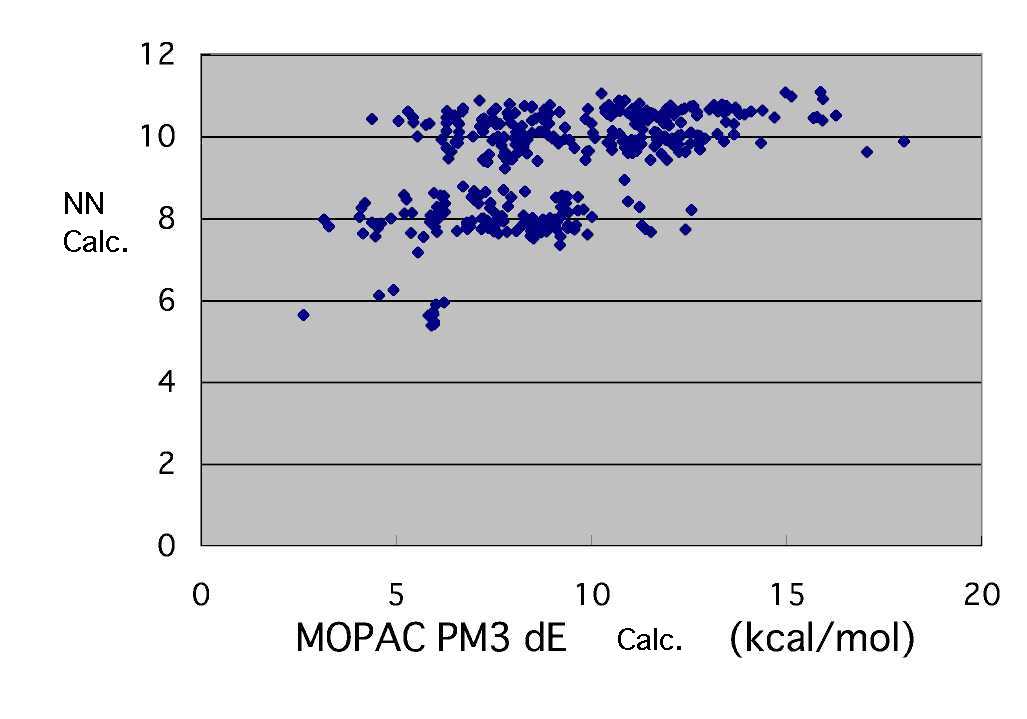
It can't be said very much that precision is still high. Do the various tuning such as to increase the number of middle layer neurons, probably a great improvement will not be carried out.
For this reaction said to decide by radical SOMO and monomer HOMO,LUMO in schoolbook, This results means some factors of other are involved.
Other factor
For this reaction said to decide by radical SOMO and monomer HOMO,LUMO in schoolbook, this results means some factors of other are involved.
For example, it is the charge of Head carbon and the radical energy level of LUMO, and the charge of Tail carbon. It is also one means to put in all the factors to invent and to narrow down by principal component analysis. Since I consider the energy level of radical LUMO should participate in this reaction, I reconstruct a neural network once again by radical SOMO, LUMO, and HOMO,LUMO of a monomer.
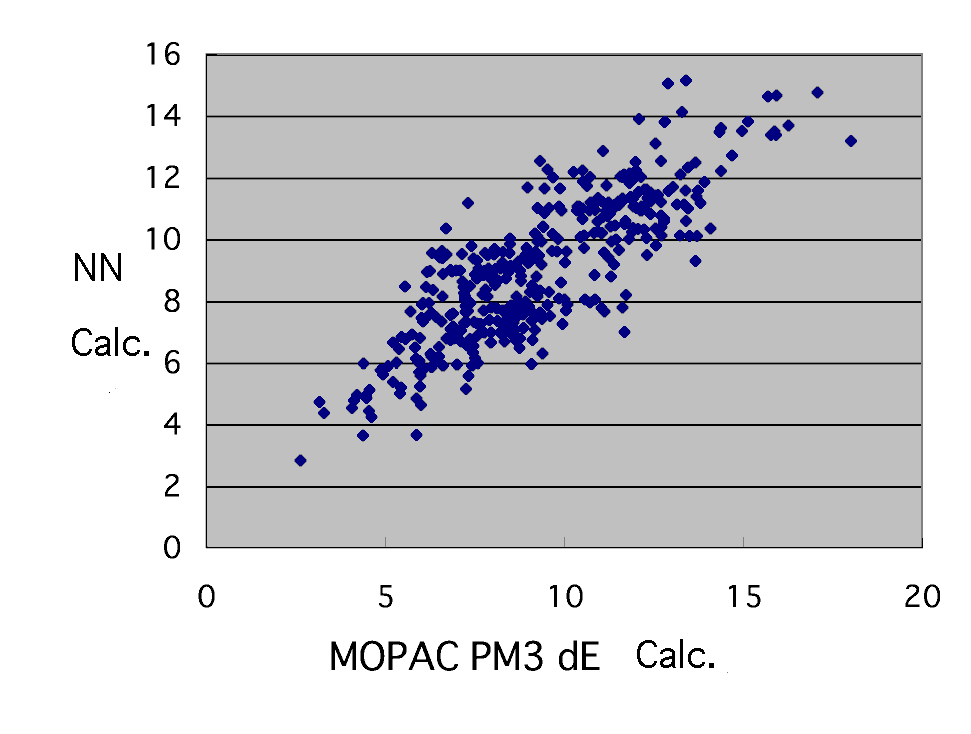
The following results were brought, when the number of the middle layer neuron was set to 5 and it learned 490000 times with the bias neuron (reconstruction learning method).
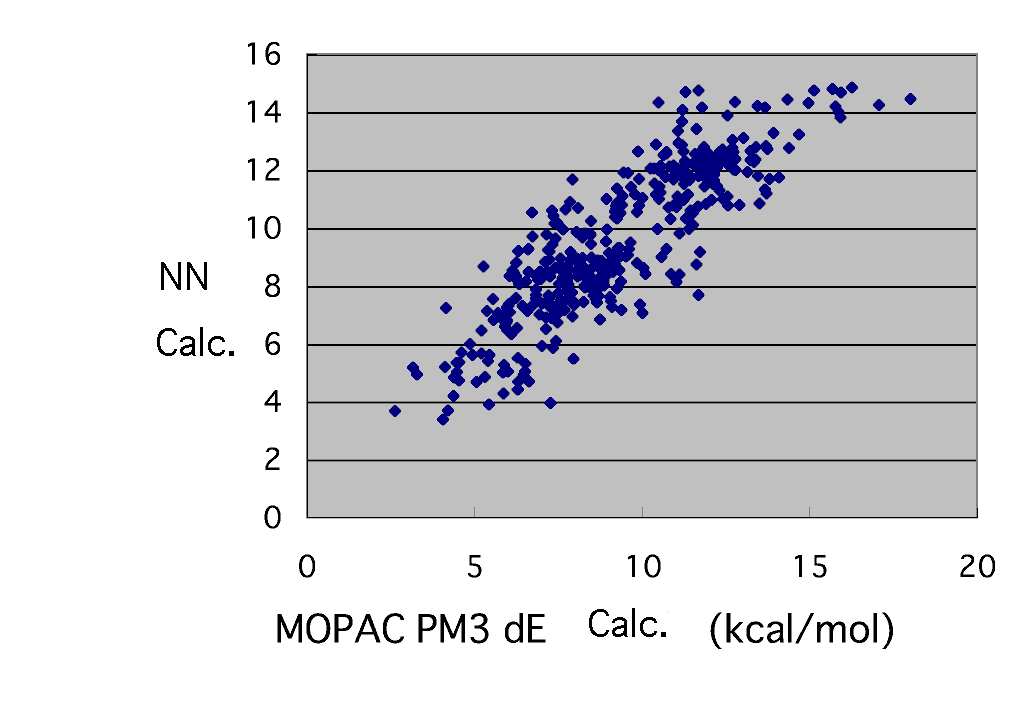
If compared with multiple regression, it turns out that it was improved fairly. Next, what should you do? A lot of plan to improve result exist, such as changing the number of middle layer neurons or putting in a charge. However, if you wants to learn the neural network method, please hold your ground just for a moment.
At first, confirm what kind of data are shifted from the straight line.
name |
dE |
NN Answer It obtains. |
Error Absolute value |
RaSOMO |
RaLUMO |
MonHOMO |
MonLUMO |
cHn-AMD |
11.68 |
7.69 |
3.99 |
-8.895 |
1.047 |
-9.794 |
0.143 |
AA-MeO |
10.48 |
14.33 |
3.85 |
-8.803 |
0.160 |
-9.515 |
1.302 |
AMD-AA |
6.70 |
10.53 |
3.83 |
-9.647 |
-0.480 |
-11.147 |
-0.170 |
AMD-MeO |
7.91 |
11.67 |
3.76 |
-9.647 |
-0.480 |
-9.515 |
1.302 |
BD-cHn |
18.01 |
14.46 |
3.55 |
-8.848 |
0.211 |
-9.593 |
1.168 |
Mal-MeO |
5.27 |
8.68 |
3.41 |
-10.963 |
-1.509 |
-9.515 |
1.302 |
MAN-MeO |
11.29 |
14.69 |
3.40 |
-9.486 |
-0.617 |
-9.515 |
1.302 |
VFR-AMD |
7.26 |
3.96 |
3.30 |
-9.343 |
0.704 |
-9.794 |
0.143 |
Vac-Mal |
7.31 |
10.61 |
3.30 |
-8.785 |
0.553 |
-11.708 |
-1.548 |
VDF-MeO |
4.15 |
7.24 |
3.09 |
-9.874 |
0.113 |
-9.515 |
1.302 |
MeO-MeO |
7.35 |
10.41 |
3.06 |
-8.352 |
1.433 |
-9.515 |
1.302 |
MMA-MeO |
11.68 |
14.74 |
3.06 |
-9.566 |
-0.598 |
-9.515 |
1.302 |
AN-MeO |
7.84 |
10.89 |
3.05 |
-9.855 |
-0.719 |
-9.515 |
1.302 |
AMD-GMA |
6.72 |
9.71 |
2.99 |
-9.647 |
-0.480 |
-10.587 |
0.143 |
VFR-cHn |
10.00 |
7.06 |
2.94 |
-9.343 |
0.704 |
-9.593 |
1.168 |
Vac-MeO |
7.71 |
10.63 |
2.92 |
-8.785 |
0.553 |
-9.515 |
1.302 |
AMD-Vac |
6.31 |
9.21 |
2.90 |
-9.647 |
-0.480 |
-10.083 |
0.687 |
cHn-AllC |
11.03 |
8.14 |
2.89 |
-8.895 |
1.047 |
-10.269 |
0.621 |
cHn-CHO |
11.62 |
8.73 |
2.89 |
-8.895 |
1.047 |
-10.681 |
-0.098 |
MAA-MeO |
11.20 |
14.07 |
2.87 |
-9.682 |
-0.717 |
-9.515 |
1.302 |
Arrange the error (|neural network Calc. - MOPAC PM3 Calc.|) in the descending order. It turns out that ten of worst 20 pieces are the system of a methyl-vinyl-ether (MeO) monomer (marked yellow). And the neural network always predicts the value high about 3kcal/mol. It means that the MeO monomer structure whose heat of formation is about 3 kcal/mol low, will exist. If the structure where transition state is high 3kcal/mol exists, it is the same. But the reaction will pass the lowest level of TS, so you need not consider higher route of TS.
So I examined the structure of methyl vinyl ether (MeO).
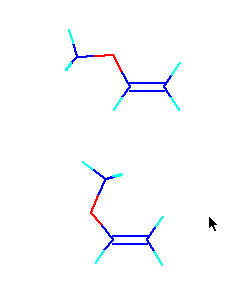
In this case, the above structures of this figure are taken. Since the surroundings of ether oxygen were free rotation, when they were turned round and the following structures were taken, they were stable 1.607kcal/mol. So, I fix the dE table of every MeO monomer. Since the value of HOMO and LUMO also changes delicately, this is all also corrected.
And a neural network is calculated once again by the newly remade data. Thus, if it corrects and calculates and corrects, calculation is repeated for a long time.
If MeO is corrected the result will become such.
name |
dE |
NN Guess value |
Absolutely Error |
somo |
lumo |
MonHOMO |
BD-cHn |
18.01 |
13.17649712 |
4.833502884 |
-8.848 |
0.211 |
-9.593 |
cHn-AMD |
11.68 |
6.985349722 |
4.694650278 |
-8.895 |
1.047 |
-9.794 |
BD-AMD |
13.66 |
9.284698015 |
4.375301985 |
-8.848 |
0.211 |
-9.794 |
Vac-Mal |
7.31 |
11.17093357 |
3.860933572 |
-8.785 |
0.553 |
-11.708 |
cHn-CHO |
11.62 |
7.787294954 |
3.832705046 |
-8.895 |
1.047 |
-10.681 |
MMA-AMD |
14.09 |
10.33804494 |
3.751955055 |
-9.566 |
-0.598 |
-9.794 |
AMD-AA |
6.7 |
10.33514547 |
3.635145471 |
-9.647 |
-0.48 |
-11.147 |
MAA-AMD |
13.71 |
10.1018309 |
3.6081691 |
-9.682 |
-0.717 |
-9.794 |
cHn-AM |
11.72 |
8.193541698 |
3.526458302 |
-8.895 |
1.047 |
-11.067 |
cHn-MMA |
11.12 |
7.658936543 |
3.461063457 |
-8.895 |
1.047 |
-10.553 |
GMA-AMD |
13.5 |
10.1030179 |
3.396982101 |
-9.699 |
-0.74 |
-9.794 |
cHn-AllC |
11.03 |
7.760870639 |
3.269129361 |
-8.895 |
1.047 |
-10.269 |
AMD-Vac |
6.31 |
9.552774897 |
3.242774897 |
-9.647 |
-0.48 |
-10.083 |
Vac-MeO |
9.311734 |
12.52751168 |
3.215777684 |
-8.785 |
0.553 |
-9.619 |
AA-AMD |
9.07 |
5.94931352 |
3.12068648 |
-10.118 |
-0.814 |
-9.794 |
BD-CHO |
13.23 |
10.1212422 |
3.108757801 |
-8.848 |
0.211 |
-10.681 |
AM-AMD |
9.38 |
6.291948181 |
3.088051819 |
-10.017 |
-0.711 |
-9.794 |
Vac-AA |
6.59 |
9.610234461 |
3.020234461 |
-8.785 |
0.553 |
-11.147 |
Vac-VDC |
5.55 |
8.46468061 |
2.91468061 |
-8.785 |
0.553 |
-9.744 |
Vac-Vac |
6.53 |
9.404180911 |
2.874180911 |
-8.785 |
0.553 |
-10.083 |
VDC-AMD |
12.31 |
9.490990594 |
2.819009406 |
-8.803 |
0.16 |
-9.794 |
And again I examined which TS was bad. The result is that the TS using the monomer of acrylamide was bad. And all of these cases, the result of NN becomes lower 3kcal/mol respectively. Supposing the result of this NN is right, The transition state should be lower.
Mathematically it is same if the heat of formation of AMD monomer is higher 3kcal/mol but higher energy structure is not stable structure, so I need not take it account.
I use MOPAC 7 and MOPAC97, but version difference will not lead 3kcal/mol .
What I found is that the position of HOMO of amide monomer mistook in this case, and it led to the calculation error.
|
|
Root No. |
12 |
13 |
14 |
15 |
16 |
|
|
|
12A |
13A |
14A |
15A |
16A |
|
|
|
-10.925 |
-10.786 |
-9.794 |
0.143 |
1.62 |
S |
C |
1 |
-0.0217 |
-0.0922 |
0.0167 |
-0.0071 |
0.2072 |
Px |
C |
1 |
0.0252 |
0.1019 |
-0.0167 |
-0.008 |
-0.0712 |
Py |
C |
1 |
-0.0574 |
-0.2712 |
0.0426 |
0.0031 |
0.186 |
Pz |
C |
1 |
0.657 |
-0.1608 |
-0.0569 |
0.5128 |
0.1986 |
S |
C |
2 |
-0.0022 |
-0.0013 |
-0.003 |
0.019 |
-0.0901 |
Px |
C |
2 |
-0.0144 |
-0.0457 |
0.006 |
-0.0186 |
0.0878 |
Py |
C |
2 |
0.0132 |
0.0701 |
-0.0134 |
-0.0036 |
0.0475 |
Pz |
C |
2 |
0.6367 |
-0.1571 |
-0.0652 |
-0.6589 |
-0.1594 |
S |
H |
3 |
0.017 |
0.0793 |
-0.0135 |
-0.0067 |
-0.0308 |
S |
H |
4 |
-0.02 |
-0.1008 |
0.0161 |
-0.0097 |
0.097 |
S |
C |
5 |
-0.0038 |
-0.0389 |
-0.0204 |
-0.0005 |
-0.3267 |
Px |
C |
5 |
-0.0086 |
-0.0792 |
0.0425 |
-0.1061 |
0.2679 |
Py |
C |
5 |
0.0225 |
0.089 |
-0.0139 |
-0.0753 |
0.1662 |
Pz |
C |
5 |
-0.0868 |
-0.0111 |
0.052 |
0.3687 |
-0.2106 |
S |
O |
6 |
0.0041 |
-0.0018 |
-0.0001 |
-0.0037 |
0.0315 |
Px |
O |
6 |
0.259 |
0.7406 |
0.015 |
0.1013 |
-0.1683 |
Py |
O |
6 |
0.0315 |
-0.3488 |
0.1241 |
0.0823 |
-0.0908 |
Pz |
O |
6 |
-0.2592 |
0.1421 |
-0.4444 |
-0.3015 |
0.138 |
S |
H |
7 |
0.0301 |
0.1215 |
-0.0138 |
-0.0194 |
-0.0008 |
S |
N |
8 |
0.0484 |
0.1695 |
0.1169 |
-0.0214 |
0.4984 |
Px |
N |
8 |
0.0941 |
0.1827 |
-0.3405 |
0.0955 |
0.0597 |
Py |
N |
8 |
0.0089 |
0.0164 |
-0.2064 |
0.0427 |
-0.0007 |
Pz |
N |
8 |
-0.0116 |
0.1789 |
0.7713 |
-0.1682 |
-0.0181 |
S |
H |
9 |
-0.0273 |
-0.1036 |
-0.0452 |
0.0268 |
-0.358 |
S |
H |
10 |
-0.0331 |
-0.0871 |
-0.0438 |
0.0165 |
-0.3695 |
The electron filled level-- RootNo14 -- therefore the energy level of LUMO ( 0.143) is O.K. Although RootNo.14( -9.794) was adopted HOMO, this was Pz orbital of nitrogen of amide (Marked yellow 0.7713). The orbital of double bond of C=C was located in the place of -10.925 ( RootNo.12). So I fixed this error.
Moreover, the monomer of lower energy found acrylic acid as well as MeO, and it also corrected this.
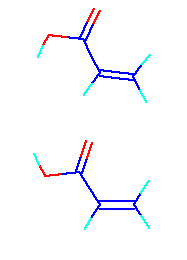
Systematic error was now corrected.
Next, Activation
energy error individually.
individual error
Systematic error was now corrected.
Next, Activation energy error individually.
Middle layer 5 |
|
|
|
|
Middle layer 4 |
|
|
|
|
Middle layer 3 |
|
|
|
name |
dE |
Answer |
d |
|
name |
dE |
Answer |
d |
|
name |
dE |
Answer |
d |
Vac-Mal |
7.31 |
10.45 |
3.14 |
|
Vac-Mal |
7.31 |
11.45 |
4.14 |
|
St-cHn |
17.07 |
11.81 |
5.26 |
VDC-MeO |
12.09 |
15.17 |
3.08 |
|
Vac-MeO |
9.31 |
13.40 |
4.09 |
|
BD-cHn |
16.90 |
12.02 |
4.88 |
AMD-Vac |
6.31 |
9.16 |
2.85 |
|
cHn-CHO |
11.62 |
7.58 |
4.04 |
|
GMA-cHn |
15.92 |
11.52 |
4.40 |
VDF-GMA |
3.16 |
5.95 |
2.79 |
|
cHn-AllC |
11.03 |
7.23 |
3.80 |
|
VFR-cHn |
10.00 |
5.77 |
4.23 |
AMD-GMA |
6.72 |
9.40 |
2.68 |
|
AN-GMA |
6.25 |
9.85 |
3.60 |
|
Mal-Vac |
5.21 |
9.16 |
3.95 |
VDF-MeO |
5.75 |
8.44 |
2.68 |
|
cHn-MMA |
11.12 |
7.52 |
3.60 |
|
MMA-cHn |
16.27 |
12.34 |
3.93 |
MeO-Mal |
7.47 |
10.09 |
2.62 |
|
Vac-GMA |
6.17 |
9.73 |
3.56 |
|
MAA-cHn |
15.78 |
11.85 |
3.93 |
AN-GMA |
6.25 |
8.74 |
2.49 |
|
AMD-GMA |
6.72 |
10.07 |
3.35 |
|
GMA-Mal |
15.87 |
12.02 |
3.85 |
VC-cHn |
12.91 |
10.43 |
2.48 |
|
cHn-AMD |
11.68 |
8.40 |
3.28 |
|
VC-cHn |
12.91 |
9.26 |
3.65 |
VDF-Vac |
3.28 |
5.76 |
2.48 |
|
AMD-Vac |
6.31 |
9.56 |
3.25 |
|
AMD-Vac |
6.31 |
9.91 |
3.60 |
Mal-MeO |
6.87 |
9.23 |
2.36 |
|
Vac-Vac |
6.53 |
9.76 |
3.23 |
|
MAN-Mal |
15.93 |
12.46 |
3.47 |
MMA-MeO |
13.28 |
15.63 |
2.35 |
|
cHn-VC |
10.72 |
7.52 |
3.20 |
|
AN-cHn |
12.41 |
8.96 |
3.45 |
BD-cHn |
16.90 |
14.55 |
2.35 |
|
AMD-Mal |
10.25 |
13.38 |
3.13 |
|
MAN-cHn |
15.69 |
12.27 |
3.42 |
AMD-MeO |
9.52 |
11.83 |
2.32 |
|
AMD-MeO |
9.52 |
12.58 |
3.07 |
|
AM-Mal |
12.56 |
9.17 |
3.39 |
VDF-VF |
4.48 |
6.76 |
2.28 |
|
cHn-VF |
10.86 |
7.82 |
3.04 |
|
Vac-VDC |
5.55 |
8.90 |
3.35 |
MMA-GMA |
10.73 |
12.97 |
2.24 |
|
VC-MeO |
9.68 |
12.65 |
2.97 |
|
AMD-GMA |
6.72 |
10.06 |
3.34 |
AMD-Mal |
10.25 |
12.47 |
2.22 |
|
cHn-AM |
11.72 |
8.77 |
2.95 |
|
Vac-GMA |
6.17 |
9.44 |
3.27 |
GMA-cHn |
15.92 |
13.70 |
2.22 |
|
Vac-VDC |
5.55 |
8.41 |
2.86 |
|
Mal-GMA |
5.97 |
9.23 |
3.26 |
St-cHn |
17.07 |
14.86 |
2.21 |
|
BD-cHn |
16.90 |
14.06 |
2.84 |
|
cHn-MeO |
12.40 |
9.27 |
3.13 |
AA-MeO |
8.98 |
11.17 |
2.18 |
|
cHn-Vac |
10.09 |
7.26 |
2.83 |
|
VDF-GMA |
3.16 |
6.16 |
3.00 |
It is the way said that the way which he does well then changes the number of a neural network's middle layer neurons, prepares a result a number of points, is investigated in an order from what has an always big error, and goes in it. In this case, the middle layer neurons was examined by 5, 4, and 3. the middle layer neurons of a with error absolute value decreases -- be alike and hang -- although it becomes large -- for example, AMD-VAc, AMD-GMA, and BD-cHn -- always -- worst one -- it enters in 20. Three, in the large thing, an error is purple and is marked yellow for two large. And regardless of such an error is large and the number of the middle layer neurons, it reexamines in an order from a bad thing.
It is a bad miss, when considering and beginning reexamination. He forgot to correct the level of SOMO of an AMD radical to the level of HOMO of an AMD monomer coming to the nitrogen atom, and having corrected. It seems that therefore, the AMD radical also had the systematically large error. The middle class is rejected once again and it is reinquiring.
Middle class 3 |
|
|
|
|
Middle class 4 |
|
|
|
|
Middle class 5 |
|
|
|
name |
dE |
Answer |
d |
|
name |
dE |
Answer |
d |
|
name |
dE |
Answer |
d |
VC-cHn |
12.91 |
8.05 |
4.86 |
|
BD-cHn |
16.90 |
12.68 |
4.22 |
|
St-cHn |
17.07 |
13.72 |
3.35 |
cHn-CHO |
11.62 |
7.13 |
4.49 |
|
AN-GMA |
6.25 |
9.89 |
3.64 |
|
Vac-Mal |
7.31 |
10.45 |
3.14 |
cHn-MeO |
12.40 |
8.05 |
4.35 |
|
AMD-cHn |
11.82 |
8.45 |
3.37 |
|
GMA-cHn |
15.92 |
13.07 |
2.85 |
BD-cHn |
16.90 |
12.68 |
4.22 |
|
BD-CHO |
13.23 |
9.86 |
3.37 |
|
BD-cHn |
16.90 |
14.08 |
2.82 |
cHn-MMA |
11.12 |
7.06 |
4.06 |
|
Vac-Mal |
7.31 |
10.29 |
2.98 |
|
MeO-Mal |
7.47 |
10.17 |
2.70 |
cHn-cHn |
11.91 |
7.92 |
3.99 |
|
BD-VC |
12.78 |
9.91 |
2.87 |
|
AM-Mal |
12.56 |
9.95 |
2.61 |
AMD-cHn |
11.82 |
7.84 |
3.98 |
|
Mal-MeO |
6.87 |
9.70 |
2.83 |
|
GMA-MAA |
13.46 |
10.85 |
2.61 |
cHn-AllC |
11.03 |
7.08 |
3.95 |
|
BD-AMD |
13.66 |
10.90 |
2.76 |
|
VDC-MeO |
12.09 |
14.68 |
2.59 |
St-cHn |
17.07 |
13.19 |
3.88 |
|
St-GMA |
9.57 |
12.31 |
2.74 |
|
MAN-CHO |
14.38 |
11.79 |
2.59 |
cHn-AMD |
11.68 |
7.89 |
3.79 |
|
AN-MeO |
9.44 |
12.17 |
2.73 |
|
MAA-cHn |
15.78 |
13.23 |
2.55 |
VDF-Vac |
3.28 |
6.97 |
3.69 |
|
VFR-GMA |
4.39 |
7.10 |
2.71 |
|
cHn-CHO |
11.62 |
9.10 |
2.52 |
MMA-cHn |
16.27 |
12.62 |
3.65 |
|
BD-MMA |
12.72 |
10.03 |
2.69 |
|
AA-VDC |
7.59 |
5.13 |
2.46 |
VDF-GMA |
3.16 |
6.80 |
3.64 |
|
AN-Vac |
6.82 |
9.46 |
2.64 |
|
AMD-cHn |
11.82 |
9.36 |
2.46 |
cHn-VC |
10.72 |
7.14 |
3.58 |
|
St-Mal |
12.55 |
15.09 |
2.54 |
|
Mal-MeO |
6.87 |
9.32 |
2.45 |
CHO-cHn |
11.52 |
8.02 |
3.50 |
|
BD-MAA |
12.29 |
9.75 |
2.54 |
|
AA-CHO |
9.18 |
6.73 |
2.45 |
MeO-cHn |
11.51 |
8.04 |
3.47 |
|
AN-MAN |
7.09 |
9.62 |
2.53 |
|
MeO-Vac |
6.34 |
8.76 |
2.42 |
cHn-VF |
10.86 |
7.41 |
3.45 |
|
St-MeO |
13.40 |
15.89 |
2.49 |
|
AA-cHn |
11.30 |
8.93 |
2.37 |
AM-cHn |
11.39 |
7.96 |
3.43 |
|
MMA-cHn |
16.27 |
13.92 |
2.35 |
|
AA-AllC |
8.65 |
6.31 |
2.34 |
AA-cHn |
11.30 |
7.93 |
3.37 |
|
GMA-cHn |
15.92 |
13.61 |
2.31 |
|
AM-cHn |
11.39 |
9.10 |
2.29 |
GMA-cHn |
15.92 |
12.57 |
3.35 |
|
cHn-CHO |
11.62 |
9.33 |
2.29 |
|
VC-cHn |
12.91 |
10.64 |
2.27 |
Three, in the large thing, an error is purple and is marked yellow for two large. In the error, the cyclohexene (cHn) monomer is involving greatly from door to door this time. And the value is wholly larger than the predicted value of the neural network(NN) method. If activation energy has shifted to the larger one, it is in the place where a transition state is lower, or is the case that the heat of formation of a monomer is higher. The neural network will think that it should have a lower transition state since a thing when the heat of formation of a monomer is higher does not need to think. Though regrettable, the ways of coping of this are not found. Having marked on the place of dE in orange expresses that in which the still lower transition state was found. For example, the mark has been carried out to 13.46 of GMA-MAA by the middle layer neurons 5.
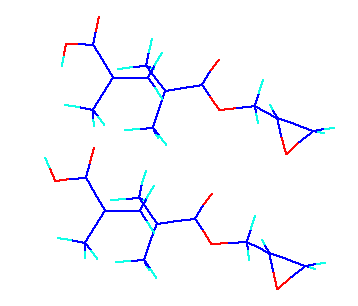
If this structure is seen, as shown in the upper figure, hydrogen of methacrylic acid has turned to the outside. If a transition state is calculated by turning an inner side for this as follows, it will become 1.5kcal /mol stability. Then, 13.46 is set to 12, and since prediction of a neural network is 10.85, what an error checks each structure to the 1.15, and is corrected corrects and goes. Most things will have improved at least mol in 1.5kcal /from 0.5, if the twist angle adjusted it bad.
When a neural network returns a big value, it is the case where there is structure with more stable monomer and radical. When the monomer (radical) is merely used in that case, only the always same quantity becomes large. When it sees roughly and there is no such tendency, it ignores for the time being.
The relative position of double combination of double combination of BD radical and a monomer, C=O of aldehyde, the spatial relationship of double combination, etc. are adjusted, and a new table is obtained.
Next, use this new table. Radical SOMO and Head carbonCharge, HOMO, LUMO, and Tail carbon charge of a monomer and Build a neural network.
neural network
use this new table. Radical SOMO and Head carbon Charge, HOMO, LUMO, and Tail carbon charge of a monomer and Build a neural network.
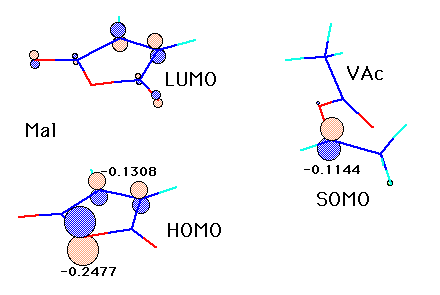
For example, when a vinyl acetate radical reacts to a maleic anhydride monomer, P orbit of the same phase as the place of double bond of maleic anhydride comes. The orbit interchanged and the knot is made of LUMO. vinyl acetate -- although there is the orbital biggest Orbit P into the portion of oxygen of maleic anhydride in lap when radical SOMO reacts -- here -- a charge ---like -- minus -- large -- Head of vinyl acetate -- it is disadvantageous that radical carbon also reacts here since a charge is minus. Then, a vinyl acetate radical reacts to double combination of maleic anhydride. Therefore, if the charge of radical Head carbon and the charge of Tail carbon of a monomer are put in and a neural network is constructed, thinking that activation energy can be guessed with more sufficient accuracy makes sense.
(As for vinylidene fluoride (VDF), in having calculated this time, only Head radical carbon is actually added.) And the activation energy of VDF-St is the minimum in 2.63 and this time. Activation energy with other monomers also brings the result that 7.96 (at the time of maleic anhydride) and which monomer react well at the maximum. If it merely does so, the radical of VDF cannot explain to itself with what whether oxygen of maleic anhydride is attacked. Although it is the thing which seems to be advantageous also in lap also in charge as for an orbit ..... strange about there is the hardness of those who have not done the molecular orbital method specially. If the direction which someone can explain comes, I will need your help.
name |
dE |
RaSOMO |
RaHCharge |
MonHOMO |
MonLUMO |
MonTCharge |
AllC-AA |
8.910044 |
-9.321 |
-0.2344 |
-11.147 |
-0.17 |
-0.0547 |
AA-MeO |
8.984206 |
-10.118 |
-0.1489 |
-9.619 |
1.25 |
-0.2969 |
MMA-MeO |
13.28327 |
-9.566 |
-0.1579 |
-9.619 |
1.25 |
-0.2969 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
AN-Vac |
6.82 |
-9.855 |
-0.0478 |
-10.083 |
0.687 |
-0.2102 |
VDF-AMD |
5.87 |
-9.874 |
0.0572 |
-10.925 |
0.143 |
-0.0608 |
AllC-GMA |
7.2 |
-9.321 |
-0.2344 |
-10.587 |
0.143 |
-0.095 |
The following results were brought, when 391 data was prepared in such a form, the number of the middle class was set to 5 and it learned 1490000 times with the bias neuron (reconstruction learning method).
Now, the result that it was worse than the case where radical SOMO, LUMO, and HOMO of a monomer and LUMO are put in, fairly was brought. If radical LUMO does not have this for an important numerical value too, activation energy is being unable to guess, even if it uses a neural network.
Then, it re-calculated by extracting what finally puts in radical SOMO, LUMO, HOMO of a monomer and LUMO, Head, and Tail carbon charge, and constructs a neural network, and a calculation result does not suit (repeatedly). 133 of 391 of the activation energy in the first table pieces were corrected after all.
The neural network finally obtained thinks that it can turn out that activation energy can be guessed only from the molecule information on a radical and a monomer very good. What it has separated from most greatly is BD-AMD. A reason is not known well.
Even if the activation energy therefore obtained by calculating a transition state by MOPAC uses a neural network, it can be guessed in the accuracy of there there. Then, how many realities were reproducible with PM3 of MOPAC primarily? I want to verify in relation to [ for photoresist ] a monomer especially.
PM3 acuracy
What calculation accuracy is it as molecular orbital calculation of MOPAC PM3? Although it may say that accuracy is not expected from bases, such as a semiempirical molecular orbital method, there are also one cup of merits, such as speed of calculation and a molecular size which can be treated. Moreover, if it is a relative value even if the absolute value of a rate of reaction is not known, it is made to compare with one r1r2 value of a radical reaction to what extent it goes.
r1 and r2 are the elementary processes of radical polymerization.
M1 and +M1-> M1- Reaction rate constant k11
M1, +M2-> M2, and k12
M2, +M1-> M1, and k21
M2, +M2-> M2, and k22
When it carries out,
It defines as r1=k11/k12 r2=k22/k21, and there are some which were collected into the polymer handbook, the textbook, etc. about various combination.
It is a reaction rate constant kij here.
kij=Aij*e(-dE/RT)
It is made the formula of Arenius to say and MOPAC and the activation energy for which it asked by PM3 are put into the place of dE. Thinking that Aij called a frequency factor will cancel oscillating calculation of MOPAC by the denominator and a molecule since it will not change a lot by the thing with low accuracy, and two kinds of monomers although originally computed from the entropy acquired from oscillating calculation, Aij ignores here. If it does so
r1=e(-dE1/RT)/e(-dE2/RT)
It will come out and the clause of temperature will also disappear. If the calculation result of PM3 is arranged, it will become like this.
PM3 |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
malR |
AMR |
VFR |
St |
1.00 |
0.05 |
0.04 |
0.26 |
0.11 |
0.44 |
0.00 |
0.03 |
0.11 |
MMA |
5.72 |
1.00 |
0.22 |
4.59 |
1.16 |
4.66 |
0.02 |
0.51 |
1.36 |
AN |
11.40 |
1.39 |
1.00 |
3.22 |
1.29 |
2.78 |
0.08 |
0.61 |
1.23 |
VDC |
3.73 |
0.28 |
0.21 |
1.00 |
0.38 |
0.37 |
0.02 |
0.13 |
0.42 |
VC |
19.12 |
0.68 |
0.59 |
2.08 |
1.00 |
4.46 |
0.03 |
0.32 |
0.99 |
Vac |
3.80 |
0.17 |
0.09 |
1.95 |
0.29 |
1.00 |
0.00 |
0.07 |
0.29 |
Mal |
28.78 |
11.39 |
16.34 |
22.74 |
5.10 |
2.18 |
1.00 |
20.83 |
4.96 |
AM |
25.05 |
3.41 |
1.45 |
8.70 |
2.21 |
13.77 |
0.23 |
1.00 |
5.19 |
VF |
15.75 |
0.66 |
0.57 |
1.86 |
1.05 |
3.52 |
0.02 |
0.34 |
1.00 |
When a styrene radical (StR) sets styrene (St) and velocity which reacts to 1 when done so for example, 5.72 can make in MMA the table which is expressing and carrying out that St tends to react 5.72 times compared with MMA.
The reference value of a reactant ratio
r1r2 |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
MalR |
AMR |
VFR |
St |
1.00 |
0.46 |
0.33 |
0.09 |
0.02 |
0.10 |
0.10 |
0.18 |
|
MMA |
0.52 |
1.00 |
0.15 |
0.25 |
0.07 |
0.02 |
0.02 |
0.34 |
|
AN |
0.40 |
1.20 |
1.00 |
0.60 |
0.04 |
0.06 |
0.00 |
0.95 |
0.005-1 |
VDC |
2.00 |
2.75 |
0.80 |
1.00 |
0.30 |
0.05 |
0.00 |
0.65 |
0.16 |
VC |
17.00 |
16.10 |
2.70 |
3.20 |
1.00 |
0.23 |
0.01 |
4.40 |
0.11 |
Vac |
55.00 |
20.00 |
4.05 |
6.70 |
1.68 |
1.00 |
0.00 |
9.00 |
0.25 |
Mal |
0.01 |
3.50 |
6.00 |
9.00 |
0.30 |
0.06 |
1.00 |
2.80 |
|
AM |
0.75 |
1.69 |
1.40 |
0.85 |
0.12 |
0.10 |
0.02 |
1.00 |
0.01 |
VF |
|
|
24-44 |
6.00 |
11.60 |
3.50 |
|
43.00 |
1.00 |
It becomes such. A reference value also changes a lot by the difference in a solvent, the difference in an initiator, and the difference among polymerization conditions. The pink color was given for the thing included in less than (1/3) 3 times. Since the percentage of victories is 20/66 noting that it is disregarded, since a diagonal clause is 1.0, it exceeds 30 percent just for a moment. Especially it returns as a general trend, it did not necessarily say that it was good and is scattered uniformly.
In the case of maleic anhydride prominent especially as alternating copolymerization, to the monomer of maleic anhydride, since a styrene radical is 0.01, by r1 of an experimental value, it reacts to maleic anhydride easily 100 times. It turns out that it turns out that it reacts, and it reacts to a styrene monomer easilier 10 times than a maleic anhydride monomer since the radical of the made maleic anhydride is 0.1, and alternating copolymerization of this system is carried out after this. In the calculation result of MOPAC PM3, since a styrene radical is 28.78, it reacts to a maleic anhydride monomer easily in the direction of a styrene monomer. Therefore, if it is concentration of the same monomer and 28-piece styrene will be connected, it is greatly shifted from reality in the result of PM3 that it returns to a styrene chain immediately since the maleic acid radical which was able to react and do one maleic anhydride is 0.00 to styrene.
Although vinyl acetate maleic anhydride is also the system of alternating copolymerization typical at r1=0.06 and r2=0.00, since it is r1=2.18, by the calculation result of PM3, it does not become alternating copolymerization.
In the transition state database, after searching for a transition state by MOPAC, it is re-calculating by B3LYP/6-31G**. When arguing about fixed-quantity nature too, in a semiempirical molecular orbital method, it is impossible. Then, it recalculated by B3LYP/6-31G**.
B3LYP |
StR |
MMAR |
ANR |
VDCR |
VCR |
VacR |
malR |
AMR |
VFR |
St |
1.00 |
0.34 |
0.20 |
1.10 |
0.16 |
13.13 |
0.00 |
0.59 |
0.03 |
MMA |
0.30 |
1.00 |
0.70 |
0.13 |
0.07 |
0.05 |
0.00 |
0.61 |
0.00 |
AN |
0.05 |
0.09 |
1.00 |
0.11 |
0.03 |
0.01 |
0.04 |
2.13 |
0.00 |
VDC |
1.22 |
1.20 |
2.42 |
1.00 |
0.24 |
0.12 |
0.02 |
2.26 |
0.02 |
VC |
8.98 |
7.16 |
9.18 |
1.66 |
1.00 |
1.01 |
0.06 |
16.86 |
0.17 |
Vac |
20.36 |
5.69 |
5.86 |
1.61 |
2.66 |
1.00 |
0.02 |
11.42 |
0.29 |
Mal |
0.03 |
0.38 |
229.14 |
4.89 |
0.08 |
0.00 |
1.00 |
2.87 |
0.00 |
AM |
0.11 |
0.30 |
0.80 |
0.11 |
0.10 |
0.11 |
0.01 |
1.00 |
0.01 |
VF |
121.45 |
14.60 |
34.43 |
2.94 |
6.33 |
7.71 |
0.06 |
25.80 |
1.00 |
It is over 60 percent by the percentages of victories 40/66 this time. A result bad at the time of the monomer of St, AN, and Mal is brought. Since it can still predict that both maleic anhydride and vinyl acetate maleic anhydride also becomes alternating copolymerization, it can be said that it is so so accuracy. Does the thing with bad calculation result in this MOPAC PM3 and coincidence of the reactant ratio r1 depend on the frequency factor having been disregarded? Or is the activation energy dE itself bad? Plot dE of PM3, and dE of B3LYP/6-31G** to a trial.
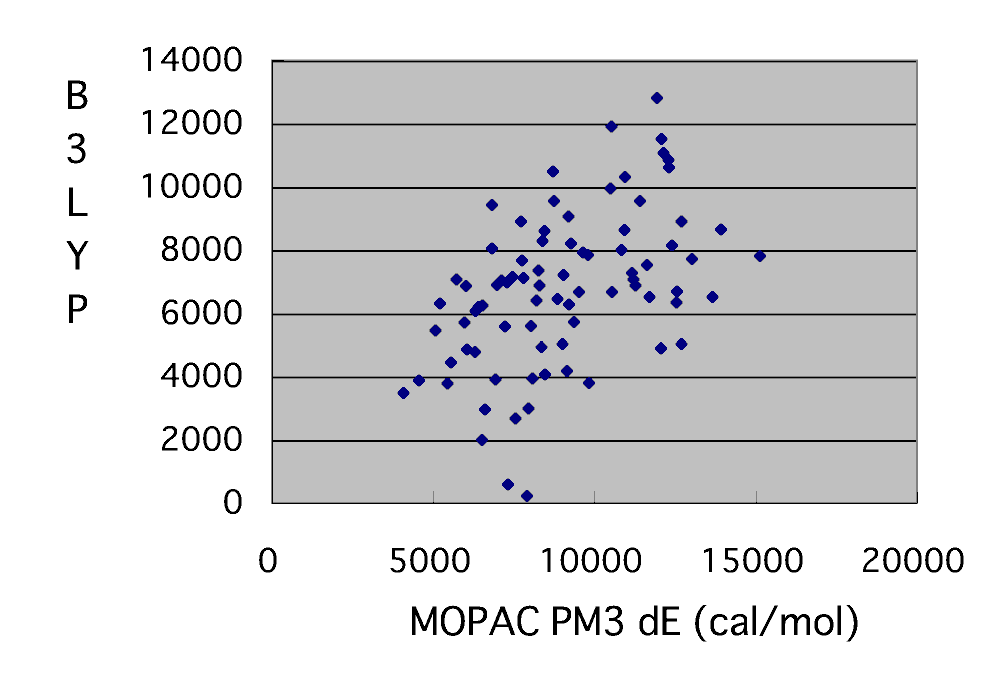
As long as this is seen, in calculation of PM3, it turns out that the dE itself is not good. It turned out that a reactant ratio is unreproducible by the cause of both that the accuracy of dE is low, and the thing for which the frequency factor was disregarded.
Then, it is in order to guess a reactant ratio by calculation. Which Level Ab Initio calculation?
| Top page Frequency View of TS Edit TS Structure How to Use Check Frequency How to use |
MOPAC Calculation Ab initio calculation Monte Carlo simulation Polymer properties estimation |
